医薬品情報
| 総称名 | オフェブ |
|---|---|
| 一般名 | ニンテダニブエタンスルホン酸塩 |
| 欧文一般名 | Nintedanib Ethanesulfonate |
| 製剤名 | ニンテダニブエタンスルホン酸塩製剤 |
| 薬効分類名 | チロシンキナーゼ阻害剤 抗線維化剤 |
| 薬効分類番号 | 3999 |
| KEGG DRUG |
D10396
ニンテダニブエタンスルホン酸塩
|
| JAPIC | 添付文書(PDF) |
添付文書情報2024年2月 改訂(第6版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| オフェブカプセル100mg | Ofev Capsules 100mg | 日本ベーリンガーインゲルハイム | 3999039M1022 | 3982.4円/カプセル | 劇薬, 処方箋医薬品注) |
| オフェブカプセル150mg | Ofev Capsules 150mg | 日本ベーリンガーインゲルハイム | 3999039M2029 | 5966.4円/カプセル | 劇薬, 処方箋医薬品注) |
1. 警告
本剤の使用は、本剤についての十分な知識と適応疾患の治療に十分な知識・経験をもつ医師のもとで行うこと。
2. 禁忌
次の患者には投与しないこと
2.1 妊婦又は妊娠している可能性のある女性[9.5参照]
2.2 本剤の成分に対し過敏症の既往歴のある患者
4. 効能または効果
5. 効能または効果に関連する注意
<全身性強皮症に伴う間質性肺疾患>
5.1 皮膚病変等の全身性強皮症に伴う間質性肺疾患以外の臓器病変に対する本剤の有効性は示されていない。
<進行性線維化を伴う間質性肺疾患>
5.2 「17.臨床成績」の項の内容を熟知し、肺機能、呼吸器症状及び胸部画像所見の総合的な評価により進行性線維化が認められる間質性肺疾患患者に本剤を投与すること。
6. 用法及び用量
通常、成人にはニンテダニブとして1回150mgを1日2回、朝・夕食後に経口投与する。なお、患者の状態によりニンテダニブとして1回100mgの1日2回投与へ減量する。
7. 用法及び用量に関連する注意
<効能共通>
7.1 下痢、悪心、嘔吐等の副作用が認められた場合は、対症療法などの適切な処置を行ったうえ、本剤の治療が可能な状態に回復するまでの間、減量又は治療の中断を検討すること。治療の中断後再開する場合は1回100mg、1日2回から再開することを検討すること。患者の状態に応じて1回150mg、1日2回へ増量することができる。再投与又は増量する場合は慎重に投与し、投与後は患者の状態を十分に観察すること。
<全身性強皮症に伴う間質性肺疾患>
7.3 シクロホスファミド、アザチオプリンとの併用時の有効性及び安全性は検討されていない。[17.1.3参照]
8. 重要な基本的注意
8.2 血小板減少があらわれ、出血に至った重篤な症例も報告されているため、本剤投与中は定期的に血液検査を行うなど、観察を十分に行うこと。[11.1.4参照]
8.3 ネフローゼ症候群があらわれることがあるので、投与期間中は尿蛋白を定期的に検査すること。[11.1.7参照]
8.4 創傷治癒を遅らせる可能性があるので、手術時は投与を中断することが望ましい。手術後の投与再開は患者の状態に応じて判断すること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 血栓塞栓症の既往歴及びその素因のある患者
血栓塞栓事象の発現を助長する可能性がある。
9.1.2 出血性素因のある患者、抗凝固剤治療を行っている患者
出血リスクを助長する可能性がある。
9.3 肝機能障害患者
9.3.1 中等度及び高度の肝機能障害(Child Pugh B、C)のある患者
9.3.2 軽度の肝機能障害(Child Pugh A)のある患者
9.4 生殖能を有する者
妊娠可能な女性に対しては、本剤の投与中及び投与終了の少なくとも3カ月後までは適切な避妊を行うよう指導すること。[9.5参照]
9.5 妊婦
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。動物実験(ラット)で乳汁中への移行が認められている。
9.7 小児等
小児等を対象とした臨床試験は実施していない。
9.8 高齢者
一般に生理機能が低下している。
10. 相互作用
相互作用序文
本剤はP-糖蛋白の基質である。
薬物代謝酵素用語
P-糖蛋白
10.2 併用注意
| P-糖蛋白阻害剤 エリスロマイシン シクロスポリン等 [16.7.1参照] | P-糖蛋白阻害剤との併用時は観察を十分に行い、異常が認められた場合は投与の中断、減量又は中止等の適切な処置を行うこと。 | P-糖蛋白の阻害により本剤の曝露が上昇する可能性がある。 ケトコナゾールとの併用によりニンテダニブのAUCが約1.6倍、Cmaxが約1.8倍に上昇した。 |
| P-糖蛋白誘導剤 リファンピシン カルバマゼピン フェニトイン セイヨウオトギリソウ(St.John's Wort、セント・ジョーンズ・ワート)含有食品等 [16.7.2参照] | P-糖蛋白誘導剤との併用により、本剤の作用が減弱する可能性がある。P-糖蛋白誘導作用のない又は少ない薬剤の選択を検討すること。 | P-糖蛋白の誘導により本剤の曝露が低下する可能性がある。 リファンピシンとの併用によりニンテダニブのAUCが約50%、Cmaxが約60%まで減少した。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 重度の下痢(3.0%)
下痢症状がみられる場合は速やかに補液やロペラミド等の止瀉剤投与を行い、本剤による治療の中断を検討すること。これらの対症療法にもかかわらず持続するような重度の下痢の場合は、本剤による治療を中止し、再投与は行わないこと。[7.1参照]
11.1.3 血栓塞栓症(静脈血栓塞栓(頻度不明)、動脈血栓塞栓(0.2%))
11.1.4 血小板減少(0.2%)
血小板減少があらわれ、出血に至った重篤な症例も報告されている。[8.2参照]
11.1.5 消化管穿孔(0.1%)
異常が認められた場合には、内視鏡、腹部X線、CT等の必要な検査を行うこと。
11.1.6 間質性肺炎(頻度不明)
胸部画像検査や呼吸機能検査で急激な悪化等の薬剤性の間質性肺炎の徴候がみられる場合は、本剤の投与を中止し、適切な処置を行うこと。
11.1.7 ネフローゼ症候群(頻度不明)[8.3参照]
11.1.8 動脈解離(頻度不明)
大動脈解離を含む動脈解離があらわれることがある1)。
11.2 その他の副作用
| 10%以上 | 5%以上10%未満 | 1%以上5%未満 | 1%未満 | |
| 代謝及び栄養障害 | 食欲減退、体重減少 | |||
| 血管障害 | 高血圧 | |||
| 胃腸障害 | 下痢(56.1%)、悪心(21.6%)、嘔吐(11.0%)、腹痛(10.9%) | 便秘 | 虚血性大腸炎 | |
| 肝胆道系障害 | 肝酵素上昇(AST、ALT、ALP、γ-GTP上昇等)(12.2%) | 高ビリルビン血症 | ||
| 皮膚及び皮下組織障害 | 発疹、そう痒症、脱毛症 | |||
| 神経障害 | 頭痛 | |||
| その他 | 出血 |
14. 適用上の注意
14.1 薬剤交付時の注意
14.1.1 PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔を起こして縦隔洞炎等の重篤な合併症を併発することが報告されている。
14.1.2 本剤は吸湿性があるので、服用直前にPTPシートから取り出すよう指導すること。また、アルミピロー包装注)のまま調剤を行うことが望ましい。
注)1アルミピロー包装中に28カプセル(14カプセル入りPTPシート×2)を含む。
14.2 薬剤投与時の注意
14.2.1 服薬を忘れた場合は、次の服薬スケジュール(朝又は夕方)から推奨用量で再開すること。
14.2.2 カプセルは噛まずにコップ一杯の水とともに服薬すること。
15. その他の注意
15.1 臨床使用に基づく情報
本剤との因果関係は明確ではないが、本剤の癌を対象とした臨床試験において顎骨壊死が認められている。また、類薬[血管内皮増殖因子受容体(VEGFR)阻害剤]において、投与後に顎骨壊死が発現したとの報告があり、多くはビスホスホネート系製剤を投与中又は投与経験のある患者であった。
15.2 非臨床試験に基づく情報
反復投与毒性試験で、ラットでは出血及び壊死を伴う切歯の破折が認められ、ラット及びサルでは、成長中の骨で骨端成長板の肥厚が認められた。
16. 薬物動態
16.1 血中濃度
日本人の特発性肺線維症患者に本剤150mg及び100mgを食後に1日2回経口投与(初回及び最終投与時は1日1回投与)した試験で得られたニンテダニブの血漿中濃度推移を図1に、薬物動態パラメータを表1に示す。ニンテダニブの血漿中濃度は投与7日目までに定常状態に達した2)。
図1 日本人の特発性肺線維症患者に本剤150mg及び100mgを食後に経口投与した試験での血漿中濃度(算術平均+標準偏差)
| パラメータ名[単位] | ||
| 初回投与後 | 150mg 11例 | 100mg 4例 |
| AUC0-12[ng・h/mL] | 152(60.6) | 59.0(67.2) |
| Cmax[ng/mL] | 34.9(62.8) | 13.2(66.9) |
| tmax[h] | 3.90(1.00-6.00) | 4.48(1.97-12.0) |
| 最終投与時 | 150mg 9例 | 100mg 4例 |
| AUCτ,ss[ng・h/mL] | 218(58.3) | 115(32.4) |
| Cmax,ss[ng/mL] | 39.7(68.1) | 20.0(64.5) |
| tmax,ss[h] | 3.87(1.00-3.97) | 3.42(2.00-4.07) |
健康成人にニンテダニブ6mgを静脈内単回投与時注)の全身クリアランスは1390mL/min、定常状態での分布容積は1050Lであった3)(外国人データ)。
全身性強皮症に伴う間質性肺疾患の患者に本剤150mgを1日2回経口投与した試験で得られたニンテダニブの定常状態時の用量補正後のトラフ血漿中濃度を表2に示す4)。
| 用量補正後トラフ血漿中濃度[ng/mL/mg] | |
| 全体集団(258例) | 0.0555(65.4) |
| 日本人集団(30例) | 0.0763(63.0) |
進行性線維化を伴う間質性肺疾患の患者に本剤150mgを1日2回経口投与した試験で得られたニンテダニブの定常状態時の用量補正後のトラフ血漿中濃度を表3に示す5)。
| 用量補正後トラフ血漿中濃度[ng/mL/mg] | |
| 全体集団(311例) | 0.0767(71.9) |
| 日本人集団(49例) | 0.107(60.5) |
16.2 吸収
16.2.1 バイオアベイラビリティ
健康成人にニンテダニブ100mgを食後に単回経口投与及び6mgを静脈内単回投与注)した結果から、絶対バイオアベイラビリティは4.69%であった3)(外国人データ)。
16.2.2 食事の影響
健康成人にニンテダニブ150mgを空腹時及び食後に単回経口投与したときの薬物動態パラメータは、表4のとおりであった6)(外国人データ)。
| パラメータ名[単位] | 空腹時 14例 | 食後 15例 |
| AUC0-∞[ng・h/mL] | 98.4(33.0)a) | 119(53.9) |
| Cmax[ng/mL] | 11.1(60.3) | 13.2(61.6) |
| tmax[h] | 2.00(1.48-3.98) | 3.98(1.50-6.05) |
16.3 分布
14C-ニンテダニブのヒト血漿蛋白結合率は97.8%であり、ヒト血液/血漿の濃度比は0.869であった7)(in vitroデータ)。
16.4 代謝
16.5 排泄
健康成人にニンテダニブ6mgを静脈内単回投与時注)の未変化体の尿中排泄率は、100mg経口投与後及び6mg静脈内投与後でそれぞれ投与量の0.05%及び1.4%であった3)(外国人データ)。
健康成人に14C-ニンテダニブ100mg溶液を単回経口投与したとき、投与放射能の0.649%が尿中に、93.4%が糞中に排泄された11)(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 肝障害患者
肝障害患者に本剤100mgを単回経口投与した場合、健康成人に比べて軽度肝障害(Child Pugh A)を有する群ではCmaxが2.2倍(90%信頼区間:1.3〜3.7)、AUCが2.2倍(90%信頼区間:1.2〜3.8)上昇し、また中等度肝障害(Child Pugh B)を有する群ではCmaxが7.6倍(90%信頼区間:4.4〜13.2)、AUCが8.7倍(90%信頼区間:5.7〜13.1)上昇した(外国人データ)。
16.6.2 高齢者
特発性肺線維症患者での母集団薬物動態解析の結果、年齢が66歳(解析対象集団の中央値)の場合に比べてAUCτは79歳では13%高くなり、52歳では14%低くなると予測された12)(日本人及び外国人の併合データ)。
16.7 薬物相互作用
16.7.1 ケトコナゾールとの併用
健康成人(31例)にケトコナゾール(P-糖蛋白阻害剤)400mgを1日1回3日間反復投与し、ケトコナゾール投与開始後3日目にニンテダニブ50mgを単回併用投与注)した場合、ニンテダニブのAUC0-∞は60.5%、Cmaxは79.6%上昇した13)(外国人データ)。
16.7.2 リファンピシンとの併用
健康成人(26例)にリファンピシン(P-糖蛋白誘導剤)600mgを1日1回7日間反復投与し、リファンピシン投与開始後8日目にニンテダニブ150mgを単回投与した場合、ニンテダニブのAUC0-∞は50.1%、Cmaxは59.8%まで低下した14)(外国人データ)。
16.7.3 ピルフェニドンとの併用
日本人の特発性肺線維症患者20例にピルフェニドンの併用/非併用下で本剤150mgを1日2回、28日間投与し、本剤及びピルフェニドンの薬物動態への影響を、それぞれ並行群間及び個体内比較で検討した。本剤をピルフェニドンと併用した場合、非併用時に比べて本剤の曝露が低くなる傾向がみられた(並行群間比較)。一方でピルフェニドンの曝露に本剤による明らかな影響は認められなかった(個体内比較)2)。
注)本剤の承認された用法・用量は1回150mg、1日2回経口投与及び1回100mg、1日2回経口投与である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
<特発性肺線維症>
17.1.1 国際共同第III相試験(1199.32試験)
特発性肺線維症患者513例(日本人55例)を対象としたプラセボ対照ランダム化二重盲検並行群間比較試験において、本剤150mg又はプラセボを1日2回、52週間経口投与した結果、主要評価項目である投与52週までの努力肺活量(FVC)の年間減少率(mL/年)は表1のとおりであり、本剤群とプラセボ群との比較において、統計学的に有意な差が認められた15)。ベースラインからのFVCの平均絶対変化量の推移を図1に示す。
| 1199.32試験 | ||
| 本剤群 | プラセボ群 | |
| FVCの年間減少率[95%信頼区間]a)(例数) | −114.7[−144.8、−84.5](309) | −239.9[−276.7、−203.1](204) |
| プラセボ群との差[95%信頼区間]a) p値a) | 125.3[77.7、172.8] p<0.0001 | / |
| ベースライン(mL) | 2756.8±735.1(309) | 2844.5±820.1(204) |
| 投与52週時(mL) | 2669.0±772.0(250) | 2664.4±834.0(165) |
| 変化量(mL) | −90.9±242.7(250) | −201.8±305.9(165) |
図1 ベースラインから投与52週時までのFVCの平均変化量の推移(平均値±標準誤差)
本試験における副作用発現割合は73.8%(228/309例)であった。主な副作用は、下痢165例(53.4%)、悪心55例(17.8%)及び食欲減退25例(8.1%)であった。
17.1.2 国際共同第III相試験(1199.34試験)
特発性肺線維症患者548例(日本人71例)を対象としたプラセボ対照ランダム化二重盲検並行群間比較試験において、本剤150mg又はプラセボを1日2回、52週間経口投与した結果、主要評価項目である投与52週までの努力肺活量(FVC)の年間減少率(mL/年)は表2のとおりであり、本剤群とプラセボ群との比較において、統計学的に有意な差が認められた16)。ベースラインからのFVCの平均絶対変化量の推移を図2に示す。
| 1199.34試験 | ||
| 本剤群 | プラセボ群 | |
| FVCの年間減少率[95%信頼区間]a)(例数) | −113.6[−144.5、−82.7](329) | −207.3[−245.3、−169.4](219) |
| プラセボ群との差[95%信頼区間]a) p値a) | 93.7[44.8、142.7] p=0.0002 | / |
| ベースライン(mL) | 2672.8±776.0(329) | 2619.0±787.3(219) |
| 投与52週時(mL) | 2637.3±811.8(269) | 2512.5±821.4(180) |
| 変化量(mL) | −86.9±283.4(269) | −204.0±280.5(180) |
図2 ベースラインから投与52週時までのFVCの平均変化量の推移(平均値±標準誤差)
本試験における副作用発現割合は69%(227/329例)であった。主な副作用は、下痢176例(53.5%)、悪心67例(20.4%)及び食欲減退29例(8.8%)であった。
<全身性強皮症に伴う間質性肺疾患>
17.1.3 国際共同第III相試験(1199.214試験)
全身性強皮症発症*から7年以内で、胸部HRCTで10%以上の線維化が認められる全身性強皮症に伴う間質性肺疾患の患者576例(日本人70例)を対象としたプラセボ対照ランダム化二重盲検並行群間比較試験において、本剤150mg又はプラセボを1日2回、52週間経口投与**した結果、主要評価項目である投与52週までの努力肺活量(FVC)の年間減少率(mL/年)は表3のとおりであり、本剤群とプラセボ群との比較において、統計学的に有意な差が認められた4)。ベースラインからのFVCの平均変化量の推移を図3に示す。
* 最初の非レイノー症状により定義
** 低用量ステロイドは、治験期間中も同一用量で併用可能とし、シクロホスファミド、アザチオプリン、中用量以上のステロイド等については併用不可と設定した。
| 1199.214試験 | ||
| 本剤群 | プラセボ群 | |
| FVCの年間減少率[95%信頼区間]a)(例数) | −52.4[−79.6、−25.2](287) | −93.3[−120.0、−66.7](288) |
| プラセボ群との差[95%信頼区間]a) p値a) | 40.95[2.88、79.01] p=0.0350 | / |
| ベースライン(mL) | 2458.5±735.9(288) | 2541.0±815.5(288) |
| 投与52週時(mL) | 2436.7±755.3(241) | 2450.3±809.4(257) |
| 変化量(mL) | −42.7±219.8(241) | −104.8±228.9(257) |
図3 ベースラインから投与52週時までのFVCの平均変化量の推移(平均値±標準誤差)
本試験における副作用発現割合は82.6%(238/288例)であった。主な副作用は、下痢197例(68.4%)、悪心71例(24.7%)及び嘔吐51例(17.7%)であった。
<進行性線維化を伴う間質性肺疾患>
17.1.4 国際共同第III相試験(1199.247試験)
進行性線維化を伴う間質性肺疾患*患者663例(日本人108例)を対象としたプラセボ対照ランダム化二重盲検並行群間比較試験において、本剤150mg又はプラセボを1日2回、52週間経口投与した。その結果、主要評価項目である投与52週までの努力肺活量(FVC)の年間減少率(mL/年)は表4のとおりであり、本剤群とプラセボ群との比較において、統計学的に有意な差が認められた。5)ベースラインからのFVCの平均変化量の推移を図4に示す。
*特発性肺線維症以外の間質性肺疾患と診断され、胸部HRCTでの線維化の広がりが肺全野の10%超で確認され、かつ医師により適切と考えられた疾患管理を行ったにもかかわらずスクリーニング前の24カ月以内において次のi)〜iv)のいずれかの間質性肺疾患の進行性の基準を満たす患者を対象とした。
i)%FVCの10%以上の減少(相対変化量)がみられる
ii)%FVCの5%以上、10%未満の減少(相対変化量)がみられ、かつ、呼吸器症状の悪化がある
iii)%FVCの5%以上、10%未満の減少(相対変化量)がみられ、かつ、胸部画像上での線維化変化の増加がみられる
iv)呼吸器症状の悪化及び胸部画像上での線維化変化の増加がみられる
| 1199.247試験 | ||
| 本剤群 | プラセボ群 | |
| FVCの年間減少率[95%信頼区間]a)(例数) | −80.8[−110.4、−51.2](332) | −187.8[−216.9、−158.6](331) |
| プラセボ群との差[95%信頼区間]a) p値a)b) | 107.0[65.4、148.5] p<0.0001 | / |
| ベースライン(mL) | 2340.1±740.2(332) | 2321.1±728.0(331) |
| 投与52週時(mL) | 2271.8±783.0(265) | 2157.8±733.0(274) |
| 変化量(mL) | −75.1±250.8(265) | −181.1±220.0(274) |
図4 ベースラインから投与52週時までのFVCの平均変化量の推移(平均値±標準誤差)
本試験の最終解析時における副作用発現割合は81.9%(272/332例)であった。主な副作用は、下痢214例(64.5%)、悪心82例(24.7%)及び嘔吐44例(13.3%)であった。
18. 薬効薬理
18.1 作用機序
18.2 抗線維化作用
ニンテダニブは、ヒト末梢血単核球を用いたin vitro試験において、線維化の発症に関与すると考えられている線維化メディエーターの放出を抑制した19)20)。さらに、ニンテダニブはin vitro試験において、PDGF、FGF及びVEGF刺激によって誘導される特発性肺線維症患者由来肺線維芽細胞の増殖及び遊走、TGF-β2によって誘導される線維芽細胞の形質転換を抑制した21)。また、全身性強皮症に伴う間質性肺疾患患者由来の肺線維芽細胞の増殖、遊走、筋線維芽細胞への形質転換及び細胞外マトリクスの発現を抑制した22)。マウス及びラットのブレオマイシン誘発肺線維症モデル、マウスのシリカ誘発肺線維症モデル、マウスの全身性強皮症に伴う間質性肺疾患モデル、及び慢性アレルゲン誘発性の肺炎症及び線維性の肺リモデリングに関するマウスモデルを用いたin vivo試験においてもニンテダニブは肺線維症に対する抗線維化効果を示した23)24)25)。
18.3 抗炎症作用
19. 有効成分に関する理化学的知見
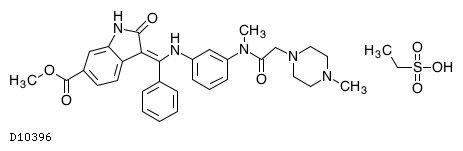
19.1. ニンテダニブエタンスルホン酸塩
| 一般的名称 | ニンテダニブエタンスルホン酸塩 |
|---|---|
| 一般的名称(欧名) | Nintedanib Ethanesulfonate |
| 化学名 | Methyl(3Z)-3-[({4-[N-methyl-2-(4-methylpiperazin-1-yl)acetamido]phenyl}amino)(phenyl)methylidene]-2-oxo-2,3-dihydro-1H-indole-6-carboxylate monoethanesulfonate |
| 分子式 | C31H33N5O4・C2H6O3S |
| 分子量 | 649.76 |
| 融点 | 305±5℃ |
| 物理化学的性状 | あざやかな黄色の粉末 |
| 分配係数 | log D=3.0(pH7.4) |
| KEGG DRUG |
|
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
<オフェブカプセル100mg>
28カプセル(14カプセル×2)PTP
<オフェブカプセル150mg>
28カプセル(14カプセル×2)PTP
23. 主要文献
- NDBを用いた調査結果の概要(VEGF/VEGFR阻害作用を有する薬剤の動脈解離に関するリスク評価), (https://www.pmda.go.jp/files/000266521.pdf)
- 社内資料:日本人患者での安全性及び薬物動態試験(2015年7月3日承認、CTD 2.7.6.2)
- 社内資料:絶対バイオアベイラビリティ試験(2015年7月3日承認、CTD 2.7.6.1)
- 社内資料:国際共同第III相試験(1199.214試験)(2019年12月20日承認、CTD 2.7.2.2、2.7.6.3)
- 社内資料:国際共同第III相試験(1199.247試験)(2020年5月29日承認、CTD 2.7.2.2、2.7.3.2、2.7.4.2、2.7.6.1)
- 社内資料:食事の影響試験(2015年7月3日承認、CTD 2.7.6.1)
- 社内資料:非臨床薬物動態試験(血漿蛋白結合)(2015年7月3日承認、CTD 2.7.2.3)
- 社内資料:非臨床薬物動態試験(代謝)(2015年7月3日承認、CTD 2.6.4.5)
- 社内資料:非臨床薬物動態試験(肝代謝)(2015年7月3日承認、CTD 2.6.4.5)
- 社内資料:非臨床薬物動態試験(小腸代謝)(2015年7月3日承認、CTD 2.6.4.5)
- 社内資料:マスバランス試験(2015年7月3日承認、CTD 2.7.6.2)
- 社内資料:特発性肺線維症患者における母集団薬物動態解析(2015年7月3日承認、CTD 2.7.2.1)
- 社内資料:ケトコナゾールとの薬物相互作用試験(2015年7月3日承認、CTD 2.7.6.2)
- 社内資料:リファンピシンとの薬物相互作用試験(2015年7月3日承認、CTD 2.7.6.2)
- 社内資料:国際共同第III相試験(1199.32試験)(2015年7月3日承認、CTD 2.7.3.3、2.7.4.2、2.7.6.3)
- 社内資料:国際共同第III相試験(1199.34試験)(2015年7月3日承認、CTD 2.7.3.3、2.7.4.2、2.7.6.3)
- Hilberg F.et al., Cancer Res., 68, 4774-4782 , (2008)
- Hilberg F.et al., J Pharmacol Exp Ther., 364, 494-503, (2018)
- 社内資料:薬効薬理試験(ヒト末梢血単核球細胞からのメディエーター放出に対する作用)(2019年12月20日承認、CTD 2.6.2.2)
- 社内資料:薬効薬理試験(ヒトT細胞からのメディエーター放出に対する作用)(2019年12月20日承認、CTD 2.6.2.2)
- Hostettler K.E.et al., Respiratory Research., 15, 157, (2014)
- 社内資料:薬効薬理試験(肺線維芽細胞の増殖,遊走及び収縮に対する作用(2019年12月20日承認、CTD 2.6.2.2)
- Huang J.et al., Ann Rheum Dis., 76, 1941-1948 , (2017)
- Wollin L.et al., J Pharmacol Exp Ther., 349, 209-220, (2014)
- 社内資料:薬効薬理試験(慢性アレルゲン誘発性の肺炎症及び線維性の肺リモデリングに関するマウスモデルにおける作用)(2020年5月29日承認、CTD 2.6.2.2)
24. 文献請求先及び問い合わせ先
文献請求先
日本ベーリンガーインゲルハイム株式会社
DIセンター
〒141-6017
東京都品川区大崎2丁目1番1号 ThinkPark Tower
電話:0120-189-779 (受付時間)9:00〜18:00(土・日・祝日・弊社休業日を除く)
製品情報問い合わせ先
日本ベーリンガーインゲルハイム株式会社
DIセンター
〒141-6017
東京都品川区大崎2丁目1番1号 ThinkPark Tower
電話:0120-189-779 (受付時間)9:00〜18:00(土・日・祝日・弊社休業日を除く)
26. 製造販売業者等
26.1 製造販売元
日本ベーリンガーインゲルハイム株式会社
東京都品川区大崎2丁目1番1号