医薬品情報
| 総称名 | ガバペン |
|---|---|
| 一般名 | ガバペンチン |
| 欧文一般名 | Gabapentin |
| 製剤名 | ガバペンチン錠 |
| 薬効分類名 | 抗てんかん剤 |
| 薬効分類番号 | 1139 |
| KEGG DRUG |
D00332
ガバペンチン
|
| KEGG DGROUP |
DG01245
ガバペンチン
|
| JAPIC | 添付文書(PDF) |
添付文書情報2026年4月 改訂(第4版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ガバペン錠200mg | GABAPEN Tablets | 富士製薬工業 | 1139007F1022 | 24.2円/錠 | 処方箋医薬品 |
| ガバペン錠300mg | GABAPEN Tablets | 富士製薬工業 | 1139007F2029 | 31円/錠 | 処方箋医薬品 |
| ガバペン錠400mg | GABAPEN Tablets | 富士製薬工業 | 1139007F3025 | 40.1円/錠 | 処方箋医薬品 |
2. 禁忌
次の患者には投与しないこと
2.1 本剤の成分に対し過敏症の既往歴のある患者
4. 効能または効果
他の抗てんかん薬で十分な効果が認められないてんかん患者の部分発作(二次性全般化発作を含む)に対する抗てんかん薬との併用療法
6. 用法及び用量
通常、成人及び13歳以上の小児にはガバペンチンとして初日1日量600mg、2日目1日量1200mgをそれぞれ3回に分割経口投与する。3日目以降は、維持量として1日量1200mg〜1800mgを3回に分割経口投与する。なお、症状により適宜増減するが、1日最高投与量は2400mgまでとする。
通常、3〜12歳の幼児及び小児にはガバペンチンとして初日1日量10mg/kg、2日目1日量20mg/kgをそれぞれ3回に分割経口投与する。3日目以降は維持量として、3〜4歳の幼児には1日量40mg/kg、5〜12歳の幼児及び小児には1日量25〜35mg/kgを3回に分割経口投与する。症状により適宜増減するが、1日最高投与量は50mg/kgまでとする。なお、いずれの時期における投与量についても、成人及び13歳以上の小児での投与量を超えないこととする。
7. 用法及び用量に関連する注意
7.1 本剤は他の抗てんかん薬と併用して使用すること。国内臨床試験において、本剤単独投与での使用経験はない。
7.2 投与初期に傾眠、ふらつき等の症状があらわれることがあるので、投与初期においては傾眠、ふらつき等の発現に十分注意しながら用量を調節すること。
7.3 1日3回投与の場合に、各投与間隔は12時間を超えないものとする。
7.4 腎機能障害のある成人患者に対する本剤の投与
腎機能障害のある成人患者に本剤を投与する場合は、下表に示すクレアチニンクリアランス値を参考として本剤の投与量及び投与間隔を調節すること。なお、ここで示している用法・用量は成人でのシミュレーション結果に基づくものであるので、腎機能低下者を対象とした国内外試験成績も踏まえて、患者ごとに慎重に観察しながら用法・用量を調節すること。[9.2.1、16.6.1参照]
| クレアチニンクリアランス(mL/min) | ≧60 | 30〜59 | 15〜29 | 5〜14 | |
| 1日投与量(mg/日) | 600〜2400 | 400〜1000 | 200〜500 | 100〜200 | |
| 投与量 | 初日 | 1回200mg 1日3回 | 1回200mg 1日2回 | 1回200mg 1日1回 | 1回200mg 1日1回 |
| 維持量 | 1回400mg 1日3回 | 1回300mg 1日2回 | 1回300mg 1日1回 | 1回300mg 2日1回 (クレアチニンクリアランスが5mL/minに近い患者では、1回200mg 2日に1回を考慮する) | |
| 1回600mg 1日3回 | 1回400mg 1日2回 | 1回400mg 1日1回 | |||
| 最高投与量 | 1回800mg 1日3回 | 1回500mg 1日2回 | 1回500mg 1日1回 | 1回200mg 1日1回 (クレアチニンクリアランスが5mL/minに近い患者では、1回300mg 2日に1回を考慮する) | |
7.5 血液透析を受けている成人患者に対する本剤の投与
血液透析を受けている成人患者に本剤を投与する際、クレアチニンクリアランスが5mL/min以上の場合には、7.4の表の投与量に加え、血液透析を実施した後に本剤200mgを追加投与する。また、クレアチニンクリアランスが5mL/min未満の場合には、初日に200mgを単回投与したのち、血液透析を実施した後に本剤1回200、300又は400mgを追加投与する(それぞれクレアチニンクリアランス60mL/min以上の患者における1回400、600又は800mg 1日3回投与に相当)。なお、ここで示している用法・用量は、48時間ごとに4時間血液透析した場合の成人でのシミュレーション結果に基づくものであるので、腎機能低下者を対象とした国内外試験成績も踏まえて、患者ごとに慎重に観察しながら用法・用量を調節すること。[9.2.2、16.6.2参照]
8. 重要な基本的注意
8.1 連用中における投与量の急激な減量ないし投与の中止により、てんかん発作の増悪又はてんかん重積状態があらわれることがあるので、投与を中止する場合には、最低1週間をかけて徐々に減量するなど慎重に行うこと。
8.2 本剤の投与により体重増加を来すことがあるので、肥満に注意し、肥満の徴候があらわれた場合は、食事療法、運動療法等の適切な処置を行うこと。特に、投与量の増加、あるいは長期投与に伴い体重増加が認められることがあるため、定期的に体重計測を実施すること。
8.3 傾眠、注意力・集中力・反射運動能力等の低下が起こることがあるので、本剤投与中の患者には自動車の運転等、危険を伴う機械の操作に従事させないよう注意すること。
8.4 本剤の投与により、弱視、視覚異常、霧視、複視等の眼障害が生じる可能性があるので、診察時に、眼障害について問診を行う等注意し、異常が認められた場合には適切な処置を行うこと。[15.2.1参照]
9. 特定の背景を有する患者に関する注意
9.2 腎機能障害患者
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。動物実験で、胎児・出生児に骨化遅延(マウス)、尿管拡張・腎盂拡張(ラット)、着床後胚死亡率の増加(ウサギ)が報告されている。
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。ヒト乳汁中へ移行することが認められている1)。
9.7 小児等
9.7.1 低出生体重児、新生児、乳児又は3歳未満の幼児を対象とした有効性及び安全性を指標とした国内臨床試験は実施していない。なお、外国で実施された3〜12歳の幼児及び小児患者を対象とした臨床試験では、本剤投与時の感情不安定、敵意、運動過多及び思考障害の発現率がプラセボ群と比較して、有意に高かったと報告されている。
9.7.2 腎機能障害のある小児患者及び透析を受けている小児患者を対象とした有効性及び安全性を指標とした臨床試験は実施していない。
9.8 高齢者
クレアチニンクリアランス値を参考に投与量、投与間隔を調節するなど慎重に投与すること。高齢者では腎機能が低下していることが多い。[16.6.4参照]
10. 相互作用
10.2 併用注意
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 急性腎障害(頻度不明)
11.1.2 皮膚粘膜眼症候群(Stevens-Johnson症候群)(頻度不明)
11.1.3 薬剤性過敏症症候群(頻度不明)
初期症状として発疹、発熱がみられ、さらに肝機能障害等の臓器障害、リンパ節腫脹、白血球増加、好酸球増多、異型リンパ球出現等を伴う遅発性の重篤な過敏症状があらわれることがある。なお、発疹、発熱、肝機能障害等の症状が再燃あるいは遷延化することがあるので注意すること。
11.1.4 肝炎(頻度不明)、肝機能障害(頻度不明)、黄疸(頻度不明)
11.1.5 横紋筋融解症(頻度不明)
筋肉痛、脱力感、CK上昇、血中及び尿中ミオグロビン上昇等があらわれた場合には、投与を中止し、適切な処置を行うこと。また、横紋筋融解症による急性腎障害の発症に注意すること。
11.1.6 アナフィラキシー(頻度不明)
アナフィラキシー(血管性浮腫、呼吸困難等)があらわれることがある。
11.2 その他の副作用
| 3%以上 | 3%未満 | 頻度不明 | |
| 精神・神経系 | 傾眠、浮動性めまい、頭痛 | 痙攣、てんかん増悪、失調、会話障害、感覚減退、記憶障害、振戦、体位性めまい、易刺激性、錯乱状態、神経過敏、不眠、不安、感情不安定、激越、攻撃性、チック | 運動障害、幻覚、ミオクローヌス、意識消失 |
| 眼 | 複視 | 眼振、眼の異常感、霧視 | 弱視、視覚異常 |
| 皮膚 | 脱毛、発疹、湿疹、じん麻疹、そう痒 | 多形紅斑 | |
| 消化器 | 悪心、嘔吐、上腹部痛、食欲減退、食欲不振、便秘、消化不良、下痢、流涎過多、食欲亢進 | ||
| 血液 | 白血球数減少、白血球数増加、ヘモグロビン減少、ヘマトクリット減少、好中球数減少、好塩基球数増加、単球数増加、好酸球数増加、血小板数減少 | ||
| 循環器 | 高血圧、動悸 | ||
| 泌尿・生殖器 | 尿失禁、尿蛋白増加、勃起機能不全 | 性欲変化、射精障害、無オルガズム症 | |
| 肝臓 | AST増加、ALT増加、Al-P増加、γ-GTP増加 | ||
| その他 | CK増加、サイロキシン減少、抗核因子陽性 | 倦怠感、関節痛、胸痛、発熱、無力症、顔面浮腫、回転性めまい、呼吸困難、背部痛、体重増加、鼻炎、耳鳴、異常歩行、LDH増加、尿酸減少、血糖増加、血糖減少、転倒・転落、鼻咽頭炎 | 血管性浮腫、浮腫、膵炎、低ナトリウム血症 |
13. 過量投与
13.1 症状
外国においてガバペンチンを49gまで経口投与した例が報告されている。過量投与後にみられた主な症状は、浮動性めまい、複視、不明瞭発語、傾眠状態、嗜眠、軽度の下痢である。
13.2 処置
これまでの例では血液透析を実施することなく回復した症例も報告されているが、本剤は血液透析により除去可能であり、発現している症状の程度に応じて血液透析の実施を考慮すること。また、重度の腎障害患者に対しても、血液透析の実施を考慮すること。[16.6.2参照]
14. 適用上の注意
14.1 薬剤交付時の注意
14.1.1 PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
14.1.2 本剤の品質は熱の影響を受けるので、高温での保存を避け、涼しいところで保存するよう指導すること。
15. その他の注意
15.1 臨床使用に基づく情報
15.1.1 海外で実施された本剤を含む複数の抗てんかん薬における、てんかん、精神疾患等を対象とした199のプラセボ対照臨床試験の検討結果において、自殺念慮及び自殺企図の発現のリスクが、抗てんかん薬の服用群でプラセボ群と比較して約2倍高く(抗てんかん薬服用群:0.43%、プラセボ群:0.24%)、抗てんかん薬の服用群では、プラセボ群と比べ1000人あたり1.9人多いと計算された(95%信頼区間:0.6-3.9)。また、てんかん患者のサブグループでは、プラセボ群と比べ1000人あたり2.4人多いと計算されている。
15.1.2 外国において、本剤投与例に原因不明の突然死が報告されている。突然死の頻度は、てんかん患者における推定値の範囲内であった。
15.1.3 臨床試験において、本剤の依存性の可能性は評価されていない。
15.2 非臨床試験に基づく情報
15.2.1 非臨床薬物動態試験において、本薬はラット、マウス、サルの水晶体に投与後10〜12時間以上にわたって分布したが、投与120時間後に水晶体から消失することがラットで確認され(マウス、サルでは消失時間を検討しなかった)、ラット及びサルの52週間反復投与毒性試験において水晶体の変化は認められなかった。眼に関する副作用の発現率はプラセボ群より有意に高く、12週間投与の国内臨床試験のプラセボ群では3.7%に対し、本剤1200mg/日群で11.6%、1800mg/日群で7.3%、長期投与では5.7%であり、12週間投与の外国臨床試験のプラセボ群では6.2%に対し、本剤600mg/日から1800mg/日投与群で9.5%から29.6%、長期投与では17.3%であった。[8.4参照]
15.2.2 がん原性試験(2年間経口投与)において、ラットの雄のみに2000mg/kg/日(最大臨床用量2400mg/日におけるヒト全身曝露量(AUC)の11倍に相当)で膵臓腺房細胞腫瘍の発生が増加したとの報告がある。雄ラットの膵臓腺房細胞腫瘍は1000mg/kg/日(最大臨床用量2400mg/日におけるヒト全身曝露量の7倍に相当)で発生の増加は認められず、また、雌のラット及び雌雄マウスでは発がん性は認められなかった。
15.2.3 幼若ラットの7週間投与試験において、雄の2000mg/kg/日で前立腺、雌の1000mg/kg/日以上で副腎の発育抑制が認められた。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
健康成人に、ガバペンチン200、400、600及び800mg(各投与量6例)を空腹時に単回経口投与した時、投与後約3時間で最高血漿中濃度に達し、消失半減期は6〜7時間であった4)。
| 投与量(mg) | Cmax(μg/mL) | AUC0-∞(μg・h/mL) | Tmax(h) | T1/2(h) |
| 200 | 2.48(21.4) | 22.64(10.3) | 3.0(30.0) | 6.47(43.0) |
| 400 | 2.94(30.8) | 27.20(27.8) | 3.1(35.5) | 6.67(27.3) |
| 600 | 4.31(16.3) | 44.12(14.4) | 3.0(20.0) | 6.13(21.9) |
| 800 | 5.23(16.6) | 52.33(17.5) | 3.3(30.3) | 6.99(25.8) |
16.1.2 反復投与
16.2 吸収
16.2.1 生物学的同等性
アジア人健康成人26例にガバペンチン200mg(シロップ又は錠)を空腹時単回投与したとき、ガバペンチンの薬物動態パラメータ及び血漿中濃度推移は以下のとおりであった。ガバペンシロップ200mgとガバペン錠200mgは生物学的に同等であることが確認された7)。
| 剤形 | Cmax(μg/mL) | AUC0-∞(μg・h/mL) | Tmax(h) | T1/2(h) |
| シロップ | 2.33(36) | 20.9(19) | 2.15(57) | 6.04(16) |
| 錠 | 2.13(27) | 21.3(16) | 2.58(47) | 5.98(17) |
| 幾何平均比a) (90%信頼区間) | 1.09 (0.98、1.22) | 0.98 (0.91、1.07) | − | − |
16.2.2 食事の影響
健康成人19例において、絶食時及び食後にガバペンチン400mgを錠剤として単回経口投与した時のCmaxはそれぞれ3.650及び3.800μg/mL、AUC0-48は35.41及び35.27μg・h/mLであった。絶食時及び食後投与後の薬物動態に差は認められなかった8)。
16.3 分布
16.3.1 健康成人12例を対象にガバペンチン150mgを静脈内単回投与した時の分布容積の平均値(変動係数%)は、57.7L(10.9)であり、ほぼ体水分量と一致した9)。ガバペンチンは血球にも移行し、血漿中濃度に対する全血中濃度の比は、0.83であった10)(外国人データ)。
16.3.2 てんかん患者において、定常状態の投与前値(トラフ値)ではガバペンチンの脳脊髄液中濃度/血漿中濃度比が約20%であった11)(外国人データ)。
16.3.3 ガバペンチンの血漿蛋白結合率は、2.0〜10.0μg/mLの血漿中濃度範囲において3%未満であった(in vitro試験)12)。
16.4 代謝
16.4.2 In vitro試験において、ガバペンチン171μg/mL(1mM、3600mg/日投与時の定常状態のCmaxの約16倍)でCYP2A6にわずかな阻害(14〜30%)が認められた。CYP1A2、2C9、2C19、2D6、2E1及び3A4に対する阻害は認められなかった14)。
16.5 排泄
16.5.1 健康成人12例を対象に、ガバペンチン150mgを静脈内単回投与した時の全身クリアランスの平均値(変動係数%)は、116.2mL/min(9.9)で、糸球体ろ過速度と一致した。この時の未変化体の尿中排泄率はほぼ100%であった9)(外国人データ)。
16.5.2 健康成人(各投与量6例)にガバペンチンを空腹時に単回経口投与した時の尿中排泄率の平均値(変動係数%)は、投与量200、400、600及び800mgで、それぞれ70.1(11.0)、42.1(30.2)、46.4(14.7)及び41.2%(15.5)であった4)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者(成人)
(1)腎機能の異なる被験者20例を対象に、ガバペンチン400mgを単回経口投与した時、腎機能の低下に従って消失半減期が延長しAUC0-∞が増加した15)(外国人データ)。
| クレアチニンクリアランス | Cmax(μg/mL) | AUC0-∞(μg・h/mL) | Tmax(h) | T1/2(h) | CLr(mL/min) |
| >60mL/min(n=6) | 3.17(28.4) | 37.8(27.4) | 4.5(18.9) | 6.5 | 81.7(32.4) |
| 30-60mL/min(n=6) | 3.52(32.2) | 73.5(31.9) | 5.1(47.1) | 12.8 | 44.7(19.7) |
| <30mL/min(n=8) | 4.93(40.5) | 551(103) | 7.1(45.6) | 52.0 | 9.0(46.9) |
(2)腎機能の異なる被験者8例(クレアチニンクリアランス:5.50〜41.4mL/min)を対象に、ガバペンチン400mgを単回経口投与し、国内健康成人男性被験者(クレアチニンクリアランス≧60mL/min)を対象とした薬物動態試験(19例)及び外国における腎機能の薬物動態に及ぼす影響を検討した試験の結果(クレアチニンクリアランス≧5mL/min、57例)と合わせて評価した。国内試験における腎機能低下者(クレアチニンクリアランス≦59mL/min)のCmaxは、外国試験と比較して高い傾向を示したが、AUC0-∞は外国試験と類似した。
CLCR:クレアチニンクリアランス
b:クレアチニンクリアランスは、24時間クレアチニンクリアランスを用いた。ただし国内健康成人男性被験者(クレアチニンクリアランス≧60mL/min)を対象とした薬物動態試験のデータに関しては、Cockcroft and Gaultの換算式を用いた。
(3)腎機能障害のある患者に投与した時の本薬の血漿中濃度シミュレーション結果
16.6.2 血液透析患者(成人)
(1)無尿症患者11例にガバペンチン400mgを単回経口投与した時、3時間の血液透析により血漿中ガバペンチン濃度は約39%減少した。その時の透析クリアランスは142mL/minであった17)(外国人データ)。
(2)週3回の血液透析を受けている日本人てんかん患者1例(CLCR=7.49mL/min)にガバペンチンを1回300mg 1日2回投与したときの血漿中ガバペンチン濃度の実測値は、母集団薬物動態モデルより算出した予測値と比較して高かった18)。
(3)血液透析を受けている患者に投与した時の本薬の血漿中濃度シミュレーション結果
16.6.3 小児
1ヵ月〜12歳の健康な小児に、ガバペンチン約10mg/kgを単回経口投与した時、5歳未満の小児におけるAUC0-∞は5歳以上と比較して約30%低かった(外国人データ)。
| 5歳未満 27例 | 5歳以上 21例 | |
| Cmax(μg/mL) | 3.74(33.5) | 4.52(26.5) |
| AUC0-∞(μg・h/mL) | 25.6(40.4) | 36.0(26.1) |
| Tmax(h) | 2.1(40.6) | 2.5(36.8) |
| T1/2(h) | 4.3(39.2) | 4.7(12.9) |
16.6.4 高齢者
16.7 薬物相互作用
16.7.1 制酸剤
16.7.2 フェニトイン
フェニトイン単剤療法中のてんかん患者8例を対象にガバペンチンを反復経口投与(1回400mg 1日3回投与)した時、ガバペンチンはフェニトインの血漿中濃度(トラフ値)に影響を及ぼさず、またフェニトインもガバペンチンの薬物動態に影響を与えなかった20)(外国人データ)。
16.7.3 カルバマゼピン
カルバマゼピン単剤療法中のてんかん患者12例を対象にガバペンチンを反復経口投与(1回400mg 1日3回投与)した時、ガバペンチンはカルバマゼピン及びその代謝物(10,11-エポキシド体)の血漿中濃度(トラフ値)に影響を及ぼさず、またカルバマゼピンもガバペンチンの薬物動態に影響を与えなかった21)(外国人データ)。
16.7.4 バルプロ酸
バルプロ酸単剤療法中のてんかん患者14例を対象にガバペンチンを反復経口投与(1回400mg 1日3回投与)した時、ガバペンチンはバルプロ酸の血清中濃度(トラフ値)に影響を及ぼさず、またバルプロ酸もガバペンチンの薬物動態に影響を与えなかった22)(外国人データ)。
16.7.5 フェノバルビタール
健康成人14例を対象にフェノバルビタール(90mg/日)及びガバペンチン(1回300mg 1日3回投与)を反復経口投与した時、ガバペンチンはフェノバルビタールの血漿中濃度(トラフ値)に影響を及ぼさず、またフェノバルビタールもガバペンチンの薬物動態に影響を与えなかった23)(外国人データ)。
16.7.6 モルヒネ
16.7.7 プロベネシド
健康成人12例を対象に、プロベネシド(1000mg単回投与)をガバペンチン投与(200mg単回投与)の1時間前に投与した時、ガバペンチンのCmax及びAUC0-∞は、ガバペンチン単独投与と比較してそれぞれ9.2%及び12.7%増加し、プロベネシドはガバペンチンの薬物動態に影響を与えなかった24)(外国人データ)。
16.7.8 シメチジン
健康成人12例を対象にシメチジン(1回300mg 1日4回投与)及びガバペンチン(400mg)を同時に単回経口投与した時、ガバペンチンのCmax及びAUC0-∞は、ガバペンチン単独投与と比較してそれぞれ6%減少及び17%増加したが、この差は臨床上問題となる差ではないと考えられた25)(外国人データ)。
16.7.9 経口避妊薬(ノルエチステロン及びエチニルエストラジオールの合剤)
健康成人女性13例を対象に経口避妊薬(ノルエチステロン2.5mg及びエチニルエストラジオール50μgの合剤1日1回投与)とガバペンチン(1回400mg 1日3回投与)を同時に経口投与した時、ガバペンチン併用時のノルエチステロンのCmax及びAUC0-24は、ガバペンチン非併用時と比較してそれぞれ13%及び3%増加し、ガバペンチンはノルエチステロンの薬物動態に影響を及ぼさなかった。また、ガバペンチン併用時のエチニルエストラジオールのCmax及びAUC0-24は、ガバペンチン非併用時と比較してそれぞれ9%及び6%増加し、ガバペンチンはエチニルエストラジオールの薬物動態に影響を及ぼさなかった26)(外国人データ)。
16.7.10 ナプロキセン
健康成人18例を対象に、ナプロキセン(250mg)及びガバペンチン(125mg)を同時に単回経口投与した時、ナプロキセンのCmax及びAUC0-∞はナプロキセン単独投与と比較してそれぞれ1%増加及び1.9%減少し、ガバペンチンはナプロキセンの薬物動態に影響を及ぼさなかった。
ナプロキセン併用時のガバペンチンのCmax及びAUC0-∞は、ガバペンチン単独投与と比較してそれぞれ14%及び12%増加し、この差は臨床上問題となる差ではないと考えられた27)(外国人データ)。
ナプロキセン併用時のガバペンチンのCmax及びAUC0-∞は、ガバペンチン単独投与と比較してそれぞれ14%及び12%増加し、この差は臨床上問題となる差ではないと考えられた27)(外国人データ)。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国内第III相試験(成人)
既存の抗てんかん薬治療では十分に抑制できない部分発作を有するてんかん患者209例を対象として、二重盲検比較試験を実施した。ガバペンチン1200mg/日、1800mg/日及びプラセボを12週間経口投与(既存の抗てんかん薬との併用)した場合、主要評価項目であるResponse Ratio注1)の評価において、いずれのガバペンチン群もプラセボ群と比較して統計的に有意な発作頻度の減少が認められた28)。
| プラセボ群 | ガバペンチン群 | ||
| 1200mg/日群 | 1800mg/日群 | ||
| 有効性評価例数 | 75 | 80 | 35 |
| Response Ratioの平均値 | −0.037 | −0.144 | −0.160 |
| 95%信頼区間 | [−0.086、0.012] | [−0.195、−0.093] | [−0.230、−0.090] |
| プラセボ群との比較(ANOVA) | p=0.0032 | p=0.0049 | |
| てんかん発作頻度減少率a) | −7.1% | −25.2% | −27.6% |
注1)Response Ratioは、本薬の投与前28日あたりの発作頻度を「B」、投与後28日あたりの発作頻度を「T」とし、(T−B)/(T+B)で算出した。その値は、−1から+1になり、0は発作頻度に変化がないこと、−1は発作が完全に消失したことを示し、正の値は発作頻度が増加したことを示す。なお、Response Ratio:−0.333は、発作頻度が50%減少したことに相当する。
a:Response Ratioの平均値から算出したてんかん発作頻度減少率(%)[=200×Response Ratio/(1−Response Ratio)]
副作用の発現率は、1200mg/日群で64.0%(55/86例)、1800mg/日群で65.9%(27/41例)であった。主な副作用は、傾眠(1200mg/日群:51.2%(44/86例)、1800mg/日群:43.9%(18/41例))、浮動性めまい(1200mg/日群:18.6%(16/86例)、1800mg/日群:19.5%(8/41例))であった。
17.1.2 国内長期投与試験(成人)
既存の抗てんかん薬治療では十分に抑制できない部分発作を有するてんかん患者211例を対象として、長期投与試験(最長200週)を実施した。評価例数は24週で170例、48週で129例、96週で55例であり、96週のガバペンチン投与において、Response Ratioの平均は−0.389〜−0.221(Response Ratioから算出したてんかん発作頻度減少率:−56.0〜−36.2%)で推移した29)30)。
副作用の発現率は、55.5%(117/211例)であった。主な副作用は、傾眠21.3%(45/211例)、浮動性めまい12.8%(27/211例)及び頭痛11.4%(24/211例)であった。
17.1.3 国内第III相試験(小児)
既存の抗てんかん薬治療では十分に抑制できない部分発作を有する3〜15歳の小児てんかん患者89例を対象として、非盲検試験を実施した。ガバペンチンを12週間経口投与した場合、主要評価項目であるResponse Ratioの平均値は主解析対象集団である86例において−0.158(Response Ratioから算出したてんかん発作頻度減少率:−27.3%)であり、発作頻度の減少が示された31)。副作用の発現率は、52.8%(47/89例)であった。主な副作用は、傾眠39.3%(35/89例)、食欲亢進3.4%(3/89例)及び痙攣3.4%(3/89例)であった。
17.1.4 国内長期投与試験(小児)
第III相試験から移行した小児てんかん患者65例を対象として、長期投与試験(52週)を実施した。長期投与試験に移行後のResponse Ratioの平均値及びResponse Ratioから算出したてんかん発作頻度減少率は下表のように推移した32)。
| 12週(開始時) | 20週 | 28週 | 36週 | 48週 | 64週 | |
| 評価例数 | 65 | 65 | 60 | 58 | 54 | 47 |
| Response Ratioの平均値 | −0.211 | −0.263 | −0.256 | −0.300 | −0.280 | −0.327 |
| てんかん発作頻度減少率b) | −34.8% | −41.7% | −40.8% | −46.1% | −43.7% | −49.3% |
副作用の発現率は、20.0%(13/65例)であった。主な副作用は、傾眠10.8%(7/65例)であった。
18. 薬効薬理
18.1 作用機序
18.2 薬理作用
18.2.1 電撃けいれんモデルにおける抗けいれん作用
ガバペンチンは、マウス及びラットにおける最大電撃による強直性伸展けいれんを用量依存的に抑制した。また、ガバペンチンはフェニトイン、カルバマゼピン及びバルプロ酸のマウスにおける最大電撃けいれん抑制作用のED50値を減少させた36)。
18.2.2 薬物誘発モデルにおける抗けいれん作用
ガバペンチンは、ペンチレンテトラゾール、ビククリン、ピクロトキシン、ストリキニーネ及びチオセミカルバジド誘発強直性伸展けいれんを抑制した。また、ペンチレンテトラゾール誘発間代性けいれんも抑制したが、ビククリン及びピクロトキシン誘発間代性けいれんを抑制しなかった36)。
18.2.3 キンドリングモデルにおける抗けいれん作用
ガバペンチンは、海馬キンドリングラットにおいて、けいれん発作行動を改善し、後発射持続時間を短縮した36)。
18.2.4 遺伝動物モデルにおける抗けいれん作用
ガバペンチンは、マウスの聴原発作及びスナネズミの反射性てんかんを抑制した。一方、ラット欠神発作(小発作)及びヒヒ光原性ミオクロニー発作には効果を示さなかった36)。
19. 有効成分に関する理化学的知見
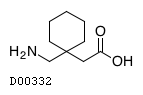
19.1. ガバペンチン
| 一般的名称 | ガバペンチン |
|---|---|
| 一般的名称(欧名) | Gabapentin |
| 化学名 | (1-Aminomethylcyclohexyl)acetic acid |
| 分子式 | C9H17NO2 |
| 分子量 | 171.24 |
| 物理化学的性状 | 白色〜微黄白色の結晶性の粉末である。 水にやや溶けやすく、エタノール(99.5)にやや溶けにくい。 |
| KEGG DRUG |
|
20. 取扱い上の注意
本剤の品質は熱の影響を受けるので、高温での保存を避け、涼しいところで保存すること。
22. 包装
<ガバペン錠200mg>
100錠[10錠(PTP)×10]
500錠[瓶]
<ガバペン錠300mg>
100錠[10錠(PTP)×10]
<ガバペン錠400mg>
100錠[10錠(PTP)×10]
23. 主要文献
- 社内資料:授乳婦における薬物動態と乳汁移行(2006年7月26日承認、CTD2.7.6)
- 社内資料:制酸剤との薬物相互作用(2006年7月26日承認、CTD2.7.2.2)
- Eckhardt,K.et al., Anesth Analg., 91 (1), 185-191, (2000) »PubMed »DOI
- 社内資料:健康成人における単回投与時の安全性と薬物体内動態(2006年7月26日承認、CTD2.7.2.2)
- 社内資料:健康成人における反復投与時の安全性と薬物動態(1800mg/日)(2006年7月26日承認、CTD2.7.2.2)
- 社内資料:健康成人における反復投与時の安全性と薬物動態(2400mg/日)(2006年7月26日承認、CTD2.7.4.2.2)
- 社内資料:生物学的同等性ならびに食事の影響(シロップ剤)(2011年7月1日承認、CTD2.7.1.2.1.1)
- 社内資料:生物学的同等性ならびに食事の影響(錠剤)(2006年7月26日承認、CTD2.7.1.2)
- 社内資料:バイオアベイラビリティ(2006年7月26日承認、CTD2.7.1.2)
- 社内資料:放射性標識体投与時の薬物動態及び代謝(2006年7月26日承認、CTD2.7.6)
- 社内資料:脳脊髄液移行及び薬物動態(2006年7月26日承認、CTD2.5.3)
- 社内資料:蛋白結合(2006年7月26日承認、CTD2.6.4.4)
- 社内資料:アンチピリンのクリアランスに対する作用(薬物代謝酵素誘導)(2006年7月26日承認、CTD2.7.6)
- 社内資料:ヒトcytochrome P450に対する阻害作用(2006年7月26日承認、CTD2.7.2.2)
- 社内資料:腎機能障害患者における薬物動態(2006年7月26日承認、CTD2.7.2.2)
- 社内資料:健康被験者、腎機能障害及びてんかん患者における母集団薬物動態(2006年7月26日承認、CTD2.7.2.3)
- 社内資料:血液透析患者における薬物動態(2006年7月26日承認、CTD2.7.2.2)
- 社内資料:腎機能低下患者における薬物動態
- 社内資料:高齢者における薬物動態(2006年7月26日承認、CTD2.7.2.2)
- 社内資料:フェニトインとの薬物相互作用(2006年7月26日承認、CTD2.7.2.2)
- 社内資料:カルバマゼピンとの薬物相互作用(2006年7月26日承認、CTD2.7.2.2)
- 社内資料:バルプロ酸との薬物相互作用(2006年7月26日承認、CTD2.7.2.2)
- 社内資料:フェノバルビタールとの薬物相互作用(2006年7月26日承認、CTD2.7.2.2)
- 社内資料:プロベネシドとの薬物相互作用(2006年7月26日承認、CTD2.7.2.2)
- 社内資料:シメチジンとの薬物相互作用(2006年7月26日承認、CTD2.7.2.2)
- 社内資料:経口避妊薬との薬物相互作用(2006年7月26日承認、CTD2.7.2.2)
- 社内資料:ナプロキセンとの薬物相互作用(2006年7月26日承認、CTD2.7.2.2)
- 社内資料:成人における二重盲検法による難治てんかん(部分発作)に対する有効性及び安全性(2006年7月26日承認、CTD2.7.6)
- 社内資料:成人における長期投与時の有効性及び安全性(第II相試験からの移行症例)(2006年7月26日承認、CTD2.7.6)
- 社内資料:成人における長期投与時の有効性及び安全性(第III相試験からの移行症例)(2006年7月26日承認、CTD2.7.6)
- 社内資料:小児における非盲検法による難治てんかん(部分発作)に対する有効性及び安全性(2006年7月26日承認、CTD2.7.3.2.1)
- 社内資料:小児における長期投与時の有効性及び安全性(2006年7月26日承認、CTD2.7.3.2.2)
- Gee,N.S.et al., J Biol Chem., 271 (10), 5768-5776, (1996) »PubMed »DOI
- Fink,K.et al., Br J Pharmacol., 130 (4), 900-906, (2000) »PubMed »DOI
- Petroff,O.A.C.et al., Epilepsia., 41 (6), 675-680, (2000) »PubMed »DOI
- 社内資料:非臨床薬理(2006年7月26日承認、CTD2.6.2.2、2.6.2.5)
24. 文献請求先及び問い合わせ先
文献請求先
富士製薬工業株式会社
くすり相談室
〒939-3515
富山県富山市水橋辻ヶ堂1515番地
電話:0120-956-792
FAX:076-478-0336
製品情報問い合わせ先
富士製薬工業株式会社
くすり相談室
〒939-3515
富山県富山市水橋辻ヶ堂1515番地
電話:0120-956-792
FAX:076-478-0336
26. 製造販売業者等
26.1 製造販売元
富士製薬工業株式会社
富山県富山市水橋辻ヶ堂1515番地