医薬品情報
| 総称名 | アリムタ |
|---|---|
| 一般名 | ペメトレキセドナトリウム水和物 |
| 欧文一般名 | Pemetrexed Sodium Hydrate |
| 製剤名 | 注射用ペメトレキセドナトリウム水和物 |
| 薬効分類名 | 代謝拮抗性抗悪性腫瘍剤 |
| 薬効分類番号 | 4229 |
| ATCコード | L01BA04 |
| KEGG DRUG |
D06503
ペメトレキセドナトリウム水和物
|
| KEGG DGROUP |
DG00682
ペメトレキセド
DG02018
代謝拮抗薬
|
| JAPIC | 添付文書(PDF) |
添付文書情報2025年7月 改訂(第3版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| アリムタ注射用100mg | Alimta Injection | 日本イーライリリー | 4229401D2026 | 18621円/瓶 | 劇薬, 処方箋医薬品注) |
| アリムタ注射用500mg | Alimta Injection | 日本イーライリリー | 4229401D1020 | 75061円/瓶 | 劇薬, 処方箋医薬品注) |
1. 警告
1.1 本剤を含むがん化学療法に際しては、緊急時に十分対応できる医療施設において、がん化学療法に十分な知識・経験を持つ医師のもとで、本剤の投与が適切と判断される症例についてのみ投与すること。適応患者の選択にあたっては、各併用薬剤の電子添文を参照して十分注意すること。また、治療開始に先立ち、患者又はその家族に有効性及び危険性を十分説明し、同意を得てから投与すること。
1.2 本剤による重篤な副作用の発現を軽減するため、必ず葉酸及びビタミンB12の投与のもとに本剤を投与すること。[7.1参照]
1.3 重度の腎機能障害患者で、本剤に起因したと考えられる死亡が報告されているので、重度の腎機能障害患者には本剤を投与しないことが望ましい。[9.2参照]
1.4 多量の胸水又は腹水が認められる患者では、体腔液の排出を検討すること。他の葉酸代謝拮抗剤で、胸水又は腹水等の体腔液の貯留が認められる患者に投与した場合、副作用の増強が報告されている。[9.1.3参照]
1.5 本剤の投与により、間質性肺炎があらわれることがあるので、本剤の投与に際しては、胸部X線検査等を行うなど観察を十分に行い、間質性肺炎が疑われた場合には、投与を中止し、適切な処置を行うこと。[8.3参照]
2. 禁忌
次の患者には投与しないこと
4. 効能または効果
5. 効能または効果に関連する注意
<効能共通>
5.1 術後補助療法における本剤の有効性及び安全性は確立していない。
<悪性胸膜中皮腫>
5.2 がん化学療法既治療例における本剤の有効性及び安全性は確立していない。
<切除不能な進行・再発の非小細胞肺癌>
5.3 扁平上皮癌等の組織型ごとの結果及び化学療法既治療例での結果を熟知し、本剤の有効性及び安全性を十分に理解した上で、患者の選択を行うこと。[17.1.5参照]
<扁平上皮癌を除く非小細胞肺癌における術前補助療法>
5.4 臨床試験に組み入れられた患者の病期等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、適応患者の選択を行うこと。[17.1.6、17.1.7参照]
6. 用法及び用量
<悪性胸膜中皮腫>
シスプラチンとの併用において、通常、成人にはペメトレキセドとして、1日1回500mg/m2(体表面積)を10分間かけて点滴静注し、少なくとも20日間休薬する。これを1コースとし、投与を繰り返す。なお、患者の状態により適宜減量する。
<切除不能な進行・再発の非小細胞肺癌>
通常、成人にはペメトレキセドとして、1日1回500mg/m2(体表面積)を10分間かけて点滴静注し、少なくとも20日間休薬する。これを1コースとし、投与を繰り返す。なお、患者の状態により適宜減量する。
<扁平上皮癌を除く非小細胞肺癌における術前補助療法>
他の抗悪性腫瘍剤との併用において、通常、成人にはペメトレキセドとして、1日1回500mg/m2(体表面積)を10分間かけて点滴静注し、少なくとも20日間休薬する。これを1コースとし、最大4コース投与を繰り返す。なお、患者の状態により適宜減量する。
7. 用法及び用量に関連する注意
<効能共通>
7.1 本剤による重篤な副作用の発現を軽減するため、以下のように葉酸及びビタミンB12を投与すること。
・葉酸
本剤初回投与の7日以上前から葉酸として1日1回0.5mgを連日経口投与する。なお、本剤の投与を中止又は終了する場合には、本剤最終投与日から22日目まで可能な限り葉酸を投与する。
・ビタミンB12
本剤初回投与の少なくとも7日前に、ビタミンB12として1回1mgを筋肉内投与する。その後、本剤投与期間中及び投与中止後22日目まで9週ごと(3コースごと)に1回投与する。[1.2参照]
7.2 欧米の添付文書中には、次表の減量基準の記載がある。
減量に関する推奨事項
次回のコース開始時の用量調節は、前回の投与コースでの最低血球数又は最大非血液毒性に基づき決定すること。回復に十分な時間をかけるために投与を延期してもよい。回復時には、表1、2、3のガイドラインに従い再投与を行うこと。これらは本剤を単剤又はシスプラチンとの併用で使用する際いずれにも適用する。
| 本剤及びシスプラチンの用量(mg/m2) | |
| 最低好中球数<500/mm3及び最低血小板数≧50,000/mm3 | 前回の用量の75% |
| 最低好中球数に関わらず最低血小板数<50,000/mm3 | 前回の用量の75% |
| 最低好中球数に関わらず出血を伴う最低血小板数<50,000/mm3 | 前回の用量の50% |
患者にグレード3以上の非血液毒性が発現した場合には、投与開始前の値以下に回復するまで本剤の投与を控えること。投与再開は表2のガイドラインに従うこと。
| 本剤の用量(mg/m2) | シスプラチンの用量(mg/m2) | |
| 粘膜炎を除くグレード3又は4の毒性 | 前回の用量の75% | 前回の用量の75% |
| 入院を要する下痢(グレードは問わない)又はグレード3若しくは4の下痢 | 前回の用量の75% | 前回の用量の75% |
| グレード3又は4の粘膜炎 | 前回の用量の50% | 前回の用量の100% |
神経毒性の発現時に推奨される本剤とシスプラチンの用量調節を表3に示す。グレード3又は4の神経毒性が認められた場合には投与を中止すること。
| CTCグレード | 本剤の用量(mg/m2) | シスプラチンの用量(mg/m2) |
| 0〜1 | 前回の用量の100% | 前回の用量の100% |
| 2 | 前回の用量の100% | 前回の用量の50% |
2回の減量後にグレード3若しくは4の血液毒性あるいは非血液毒性が認められた場合又はグレード3若しくは4の神経毒性が観察された場合は直ちに本剤の投与を中止すること。
<悪性胸膜中皮腫>
7.3 シスプラチンは本剤投与30分後に75mg/m2(体表面積)を投与し、投与に際しては、シスプラチンの電子添文に従い腎毒性軽減のための処置等を行うこと。
7.4 切除不能な進行・再発の悪性胸膜中皮腫に対してペムブロリズマブ(遺伝子組換え)及びカルボプラチンと併用する際の用法及び用量は、ペムブロリズマブ(遺伝子組換え)の電子添文を参照すること。
7.5 本剤を単剤で使用した場合の有効性及び安全性は確立していない。
<扁平上皮癌を除く非小細胞肺癌における術前補助療法>
7.6 本剤の投与回数及び併用する他の抗悪性腫瘍剤について、「17.臨床成績」の項の内容を熟知し、関連学会の最新のガイドライン等を参考にした上で選択すること。[17.1.6、17.1.7参照]
8. 重要な基本的注意
8.2 骨髄抑制等の重篤な副作用が起こることがあるので、投与に際しては臨床症状を十分に観察し、頻回に臨床検査(血液学的検査、肝機能検査、腎機能検査等)を行うこと。また、本剤の投与にあたっては、G-CSF製剤の適切な使用に関しても考慮すること。[9.1.1、11.1.1参照]
8.3 間質性肺炎等の重篤な肺毒性が起こることがあるので、本剤の投与にあたっては、臨床症状(呼吸状態、咳及び発熱等の有無)を十分に観察し、定期的に胸部X線検査を行うこと。また、必要に応じて胸部CT検査、動脈血酸素分圧(PaO2)、肺胞気動脈血酸素分圧較差(A-aDO2)、肺拡散能力(DLco)等の検査を行い、患者の状態を十分に観察すること。[1.5、9.1.2、11.1.3参照]
8.4 重度の腎機能障害患者で、本剤に起因したと考えられる死亡が報告されているので、本剤投与前に患者の腎機能を確認すること。[9.2.1参照]
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 骨髄抑制のある患者[8.2参照]
9.1.2 間質性肺炎、肺線維症、又はこれらの疾患の既往歴のある患者[8.3参照]
9.1.3 胸水又は腹水が認められる患者
多量の体腔液が認められる患者では、本剤投与前に体腔液の排出を検討すること。胸水、腹水等体腔液の本剤投与への影響は不明であるが、他の葉酸代謝拮抗剤で副作用の増強が報告されている。[1.4参照]
9.2 腎機能障害患者
本剤は主として腎より排泄される。腎機能障害の程度に応じて本剤の血中濃度の増加が認められている。クレアチニンクリアランスが45mL/min未満の患者は臨床試験では除外されている。[1.3参照]
9.2.1 重度の腎機能障害患者[8.4参照]
9.3 肝機能障害患者
臨床試験では除外されている。
9.4 生殖能を有する者
9.4.1 生殖可能な年齢の患者に投与する必要がある場合には、性腺に対する影響を考慮すること。動物実験で雄性生殖器に対する影響(マウス:精子形成能の低下あるいは精細管変性、イヌ:精細管上皮の変性あるいは壊死)が報告されている。
9.4.2 妊娠する可能性のある女性には、本剤投与中及び最終投与後6ヵ月間において避妊する必要性及び適切な避妊法について説明すること。[9.5、15.2参照]
9.4.3 男性には、本剤投与中及び最終投与後3ヵ月間においてバリア法(コンドーム)を用いて避妊する必要性について説明すること。[9.5、15.2参照]
9.5 妊婦
9.6 授乳婦
授乳しないことが望ましい。乳汁中への移行については不明である。
9.7 小児等
小児等を対象とした臨床試験は実施していない。
9.8 高齢者
患者の状態を観察しながら慎重に投与すること。一般に生理機能が低下していることが多い。
10. 相互作用
10.2 併用注意
| 非ステロイド性抗炎症剤 イブプロフェン等 | 本剤の血中濃度が増加し、副作用が増強するおそれがあるので、併用療法を行う場合には、頻回に臨床検査を行うなど患者の状態を十分に観察すること。 | 他の葉酸代謝拮抗剤で副作用の増強が知られており、本剤においてもクリアランスの低下が認められている。 |
| 腎毒性を有する薬剤又は腎排泄型薬剤 プロベネシド、ペニシリン等 | 本剤の血中濃度が増加し、副作用が増強するおそれがあるので、併用療法を行う場合には、慎重に投与すること。 | 他の葉酸代謝拮抗剤で腎排泄を競合的に阻害することが知られており、本剤のクリアランスを遅延させるおそれがある。 |
| 抗悪性腫瘍剤 | 骨髄機能抑制等の副作用が増強するおそれがあるので、併用療法を行う場合には、患者の状態を十分に観察すること。 | ともに骨髄機能抑制作用を有する。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 骨髄抑制
白血球減少(71.6%)、好中球減少(64.4%)、ヘモグロビン減少(54.2%)、リンパ球減少(51.1%)、血小板減少(46.2%)、貧血(頻度不明)、発熱性好中球減少(頻度不明)、汎血球減少症(頻度不明)があらわれることがある。[8.2参照]
11.1.2 感染症
敗血症(頻度不明)、肺炎(頻度不明)等の重篤な感染症があらわれることがある。
11.1.3 間質性肺炎(3.6%)
肺毒性の発症あるいは急性増悪が疑われた場合には、直ちに本剤による治療を中止し、ステロイド治療等の適切な処置を行うこと。[8.3参照]
11.1.4 ショック(頻度不明)、アナフィラキシー(頻度不明)
呼吸困難、喘鳴、血圧低下、発疹、発赤、そう痒感等の異常が認められた場合には投与を中止し、適切な処置を行うこと。
11.1.5 重度の下痢(1.3%)
11.1.6 脱水(1.3%)
異常が認められた場合には、減量、休薬、補液、電解質投与等適切な処置を行うこと。
11.1.7 腎不全
クレアチニン上昇(7.1%)、腎不全(頻度不明)、クレアチニンクリアランス低下(頻度不明)があらわれることがある。
11.1.8 中毒性表皮壊死融解症(Toxic Epidermal Necrolysis:TEN)(頻度不明)、皮膚粘膜眼症候群(Stevens-Johnson症候群)(頻度不明)
11.2 その他の副作用
| 20%以上 | 5〜20%未満 | 5%未満 | 頻度不明 | |
| 内分泌系 | 血糖値上昇 | 尿糖陽性 | ||
| 精神神経系 | 頭痛、めまい、感覚神経障害 | 味覚異常、感覚鈍麻、不眠症、傾眠、運動神経障害 | ||
| 眼 | 眼脂、流涙増加、眼球乾燥、結膜炎 | |||
| 循環器 | 血圧上昇、心嚢液貯留、動悸、不整脈 | |||
| 血管障害 | ほてり | 潮紅 | ||
| 呼吸器 | しゃっくり、咳嗽、咽喉頭疼痛、鼻漏、呼吸困難、胸水、低酸素症 | |||
| 消化器 | 食欲不振、悪心、嘔吐 | 便秘、下痢、口内炎・咽頭粘膜炎、消化不良 | 口唇炎、胃部不快感、腹痛、胃炎、食道炎 | 大腸炎 |
| 肝臓 | AST上昇、ALT上昇、血中LDH上昇、血中Al-P上昇 | ビリルビン上昇、γ-GTP上昇 | 尿中ウロビリン陽性 | |
| 皮膚 | 発疹 | そう痒症 | 色素沈着、脱毛症、多形紅斑、蕁麻疹 | |
| 腎臓 | アルブミン低下、電解質異常、尿潜血陽性、蛋白尿、総蛋白減少、BUN上昇 | 総蛋白増加 | ||
| その他 | 倦怠感、発熱、CRP上昇 | 疲労、体重減少、熱感、白血球増多、好中球増多、血小板増多、浮腫 | 関節痛、感冒様症状、顔面浮腫、眼瞼浮腫、悪寒、鼻出血、肺炎、単球増多、胸痛、アレルギー反応/過敏症 | 放射線照射リコール反応、溶血性貧血 |
13. 過量投与
13.1 症状
主な症状は、骨髄抑制(好中球減少、血小板減少、貧血)、粘膜炎及び発疹である。また、感染及び下痢があらわれることがある。
13.2 処置
症状に応じた支持療法を行う他、ホリナートカルシウムによる処置を検討すること。
14. 適用上の注意
14.1 薬剤調製時の注意
14.1.1 本剤は細胞毒性を有するため、調製時には手袋を着用することが望ましい。皮膚に薬液が付着した場合は直ちに石けんでよく洗浄し、粘膜に付着した場合は直ちに多量の流水でよく洗い流すこと。
14.1.2 本剤の溶解及び希釈には日局生理食塩液のみを使用すること。カルシウムを含有する溶液との混合により濁り又は沈殿が確認されているので、乳酸リンゲル液及びリンゲル液等との配合を避けること。また、他剤との混注を行わないこと。
14.1.3 本剤1バイアルに日局生理食塩液を、アリムタ注射用100mgの場合4.2mL、アリムタ注射用500mgの場合20mLを注入して十分に溶解する。溶解後のペメトレキセド濃度は25mg/mL(実測値)である。投与量に応じて必要量の溶解液を抜き取り、日局生理食塩液に混和して100mLとして用いる。
14.1.4 溶解後は速やかに投与すること。保存する場合は冷蔵(2〜8℃)にて保存し、24時間以内に使用すること。溶解した残液は使用しないこと。
14.2 薬剤投与時の注意
必ず点滴静脈内投与とし、皮下、筋肉内には投与しないこと。
15. その他の注意
16. 薬物動態
16.1 血中濃度
16.1.1 血漿中濃度
各種悪性腫瘍患者31例に本剤を300〜1,200mg/m2注)の用量範囲で21日ごとに10分間点滴静注した。血漿中濃度は点滴終了直後が最も高く、その後速やかに消失し、消失半減期は2.74時間(範囲:2.28〜3.62時間)であった。このときの血漿クリアランスは61.4〜109mL/min、定常状態分布容積は10.6〜14.8Lであった。第1コース及び第2コース間で血漿中濃度に差は認められなかった1)。
図1)ペメトレキセド投与後の血漿中濃度推移(平均±標準偏差)
16.3 分布
16.3.1 組織分布
(参考)
マウスに14C標識体20mg/kgを単回静注したとき、肺等の広範な臓器・組織に速やかに分布した。投与1時間後には、尿、胆のう内胆汁、糞、腸内容物、腎臓及び肝臓に比較的高い放射活性が検出され、本剤が投与後速やかに尿中及び胆汁中に排泄されることが示唆された2)。
16.3.2 蛋白結合率
本剤のヒト蛋白結合率は約81%であった。また、本剤のヒト蛋白結合率は腎機能障害による影響をほとんど受けなかった3)(in vitro)。
16.4 代謝
本剤は主として尿中へ未変化体として排泄されることから1)、代謝をほとんど受けないと推察された。
16.5 排泄
各種悪性腫瘍患者31例に本剤を300〜1,200mg/m2注)の用量範囲で21日ごとに10分間点滴静注した。本剤は点滴静注後24時間以内に、その大部分が主に尿中へ未変化体として排泄され、投与後72時間までの累積尿中未変化体排泄率は75.2%(64.5〜82.7%)であった1)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
日本人患者31例と外国人患者412例の統合解析により、本剤の薬物動態に与える腎機能の影響を評価した。日本人の腎機能低下患者(クレアチニンクリアランス45mL/min)に本剤500mg/m2を投与した場合、腎機能が正常な患者(クレアチニンクリアランス90mL/min)に比較して、本剤の血漿クリアランスが32%低く、血漿中濃度時間曲線下面積(AUC)が48%増大すると予測された4)。
16.7 薬物相互作用
葉酸とビタミンB12の併用は、本剤の単剤投与時、あるいはシスプラチンとの併用投与時とも本剤の血漿クリアランスに影響を与えないことが示された。また、本剤とシスプラチンは互いの薬物動態に影響を及ぼさないことが明らかとなった5)(外国人データ)。
注)本剤の承認された1回用量は、500mg/m2(体表面積)である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
<悪性胸膜中皮腫>
17.1.1 国内第I/II相試験
悪性胸膜中皮腫患者を対象に国内で実施した併用投与第I/II相試験注19)において、本剤500mg/m2及びシスプラチン75mg/m2を投与された症例の奏効率は36.8%(19例中PR7例)であった。
本治療との因果関係を否定できない死亡例が全投与症例25例中1例に認められた。安全性評価対象25例中に認められた主な副作用は、悪心(96.0%)、ヘモグロビン減少(96.0%)、食欲不振(88.0%)、好中球減少(84.0%)、赤血球減少(84.0%)、白血球減少(80.0%)、嘔吐(72.0%)、リンパ球減少(64.0%)、倦怠感(56.0%)、血中尿素増加(52.0%)であった。[8.1参照]
17.1.2 化学療法未治療患者を対象とした外国第III相試験
悪性胸膜中皮腫患者(化学療法未治療)を対象に米国ほか20ヵ国で実施された第III相試験注19)における、本剤500mg/m2及びシスプラチン併用投与群75mg/m2及びシスプラチン75mg/m2単独投与群(未承認)注1)の成績は、次表のとおりであった6)。なお、本試験は優越性を検証することを主要目的として実施した。
| 本剤及びシスプラチン併用投与群 | シスプラチン単独投与群注1) | |
| N注2) | 226 | 222 |
| 生存期間中央値(月) | 12.1 | 9.3 |
| p値=0.020注3) | ||
注1)シスプラチン単独投与群(未承認):21日を1コースとして第1日目に、シスプラチン75mg/m2を投与
注2)薬剤を投与された症例(葉酸、ビタミンB12の併用なし症例を含む)
注3)ログランク検定(優越性に関する検定)
本剤とシスプラチンの併用投与群において、本治療との因果関係を否定できない死亡例が全投与症例226例中3例に認められ、いずれも葉酸及びビタミンB12が併用投与されていない症例であった。安全性評価対象168例(葉酸及びビタミンB12併用群)中に認められた主な副作用は、悪心(82.1%)、嘔吐(56.5%)、好中球減少(56.0%)、白血球減少(53.0%)、疲労(47.6%)、ヘモグロビン減少(26.2%)、血小板減少(23.2%)、口内炎(23.2%)、食欲不振(20.2%)であった。[8.1参照]
<切除不能な進行・再発の非小細胞肺癌>
17.1.3 化学療法既治療患者を対象とした国内第II相試験
非小細胞肺癌患者(化学療法既治療)を対象に国内で実施した第II相試験注19)において、本剤500mg/m2を投与された症例の奏効率は18.5%(108例中PR20例)であった。
本剤(500mg/m2又は1,000mg/m2注4)投与)との因果関係を否定できない死亡例が全投与症例226例中1例に認められた。安全性評価対象225例中に認められた主な副作用は、AST上昇(76.9%)、発疹(73.8%)、白血球減少(71.6%)、ALT上昇(71.6%)、好中球減少(64.4%)、食欲不振(56.9%)、ヘモグロビン減少(54.2%)、悪心(53.8%)、LDH上昇(52.0%)、リンパ球減少(51.1%)であった。[8.1参照]
注4)本剤の承認された1回用量は、500mg/m2(体表面積)である。
17.1.4 化学療法未治療患者を対象とした外国第III相試験
非小細胞肺癌患者(化学療法未治療)を対象に米国ほか26ヵ国で実施された第III相試験注19)における、本剤500mg/m2及びシスプラチン75mg/m2併用投与群とゲムシタビン1250mg/m2及びシスプラチン75mg/m2併用投与群の成績は、次表及び図のとおりであった7)。なお、本試験は非劣性を検証することを主要目的として実施した。
| 本剤及びシスプラチン併用投与群注5) | ゲムシタビン及びシスプラチン併用投与群注6) | |
| N注7) | 862 | 863 |
| 生存期間中央値(月) (95%信頼区間) | 10.3 (9.8-11.2) | 10.3 (9.6-10.9) |
| ハザード比 (95%信頼区間) | 0.94(0.84-1.05)注8) p値<0.0001注9) | |
図1)外国第III相試験における非小細胞肺癌患者(化学療法未治療)に対する生存率の推移
AC群:本剤及びシスプラチン併用投与群
GC群:ゲムシタビン及びシスプラチン併用投与群
本試験における組織型別の部分集団解析の結果を以下の表及び図に示す8)。
| 組織型別部分集団 | 生存期間中央値(月) (95%信頼区間) | ハザード比注10),注11) (95%信頼区間) | |||
| 本剤及びシスプラチン併用投与群 | ゲムシタビン及びシスプラチン併用投与群 | ||||
| 扁平上皮癌(N=473) | 9.4 (8.4-10.2) | N=244 | 10.8 (9.5-12.1) | N=229 | 1.23 (1.00-1.51) |
| 腺癌(N=847) | 12.6 (10.7-13.6) | N=436 | 10.9 (10.2-11.9) | N=411 | 0.84 (0.71-0.99) |
| 大細胞癌(N=153) | 10.4 (8.6-14.1) | N=76 | 6.7 (5.5-9.0) | N=77 | 0.67 (0.48-0.96) |
| その他注12)(N=252) | 8.6 (6.8-10.2) | N=106 | 9.2 (8.1-10.6) | N=146 | 1.08 (0.81-1.45) |
図2)外国第III相試験における非小細胞肺癌患者(化学療法未治療)に対する組織型別生存率の推移
AC群:本剤及びシスプラチン併用投与群
GC群:ゲムシタビン及びシスプラチン併用投与群
本剤とシスプラチンの併用群839例において、本治療との因果関係を否定できない有害事象は、悪心(56.1%)、嘔吐(39.7%)、ヘモグロビン減少(33.0%)、疲労(42.7%)、好中球減少症(29.0%)であった。[8.1参照]
17.1.5 化学療法既治療患者を対象とした外国第III相試験
非小細胞肺癌患者(化学療法既治療)を対象に米国ほか23ヵ国で実施された第III相試験注19)における、本剤500mg/m2投与群及びドセタキセル75mg/m2投与群注13)の成績は、次表及び図のとおりであった9)。なお、本試験は非劣性を検証することを主要目的として実施した。
| 本剤投与群 | ドセタキセル投与群注13) | |
| N注14) | 283 | 288 |
| 生存期間中央値(月) (95%信頼区間) | 8.3 (7.0-9.4) | 7.9 (6.3-9.2) |
| ハザード比 (95%信頼区間) | 0.99(0.82-1.20) p値=0.251注15) | |
注13)ドセタキセル投与群:21日を1コースとして第1日目に、ドセタキセル75mg/m2を投与
注14)すべての無作為割付された症例
注15)ワルド検定(非劣性に関する検定)
図3)外国第III相試験における非小細胞肺癌患者(化学療法既治療)に対する生存率の推移
A群:本剤投与群
D群:ドセタキセル投与群
本試験における組織型別の部分集団解析の結果を以下の表及び図に示す8)。
| 組織型別部分集団 | 生存期間中央値(月) (95%信頼区間) | ハザード比注16),注17) (95%信頼区間) | |||
| 本剤投与群 | ドセタキセル群 | ||||
| 扁平上皮癌(N=172) | 6.2 (4.9-8.0) | N=78 | 7.4 (5.6-9.5) | N=94 | 1.56 (1.08-2.26) |
| 腺癌(N=301) | 9.0 (7.6-9.6) | N=158 | 9.2 (7.5-11.3) | N=143 | 0.92 (0.69-1.22) |
| 大細胞癌(N=47) | 12.8 (5.8-14.0) | N=18 | 4.5 (2.3-9.1) | N=29 | 0.27 (0.11-0.63) |
| その他注18)(N=51) | 9.4 (6.0-10.1) | N=29 | 7.9 (4.0-8.9) | N=22 | 0.57 (0.27-1.20) |
図4)外国第III相試験における非小細胞肺癌患者(化学療法既治療)に対する組織型別生存率の推移
A群:本剤投与群
D群:ドセタキセル投与群
注19)発疹の発現及び重症化を軽減するため、外国臨床試験では、本剤投与の前日から投与の翌日までの3日間、デキサメタゾンを1回4mg、1日2回経口投与した。また、国内臨床試験では、発疹が発現した症例に限り、次回の本剤投与時から外国臨床試験の用法・用量を参考にデキサメタゾン等の副腎皮質ホルモン剤の投与を可能とした。[8.1参照]
<扁平上皮癌を除く非小細胞肺癌における術前補助療法>
17.1.6 国際共同第III相試験(ONO-4538-55/CA209816試験)
臨床病期IB(腫瘍径が4cm以上)、II又はIIIAの非小細胞肺癌の術前患者注20)358例(日本人患者68例を含む。ニボルマブ(遺伝子組換え)と白金系抗悪性腫瘍剤を含む化学療法併用(N+C併用)注21)群179例、白金系抗悪性腫瘍剤を含む化学療法群179例)を対象に、化学療法を対照として、N+C併用の有効性及び安全性を検討した。主要評価項目の一つである無イベント生存期間(中央値[95%信頼区間])は、N+C併用群で31.57[30.16〜推定不能]ヵ月、化学療法群で20.80[14.03〜26.71]ヵ月であり、N+C併用投与は化学療法に対し統計学的に有意な延長を示した(ハザード比0.63[97.38%信頼区間:0.43〜0.91]、p=0.0052[層別log-rank検定]、2021年9月8日データカットオフ)。
図5)国際共同第III相試験における非小細胞肺癌の術前患者に対する無イベント生存率の推移
ニボルマブ(遺伝子組換え)、ペメトレキセド及びシスプラチン注21)が併用投与された患者における安全性評価対象83例中61例(73.5%)に副作用が認められた。主な副作用は、悪心30例(36.1%)、便秘21例(25.3%)、食欲減退15例(18.1%)、倦怠感10例(12.0%)、発疹10例(12.0%)、好中球減少症9例(10.8%)、無力症9例(10.8%)であった(2022年9月6日データカットオフ)。[5.4、7.6参照]
注20)臨床病期はAmerican Joint Committee on Cancer(AJCC)/Union for International Cancer Control(UICC)病期分類(第7版)に基づく。
EGFR遺伝子変異陽性又はALK融合遺伝子陽性であることが確認されている患者は対象外とされた。
EGFR遺伝子変異陽性又はALK融合遺伝子陽性であることが確認されている患者は対象外とされた。
注21)扁平上皮癌に対しては、ニボルマブ(遺伝子組換え)1回360mg、ゲムシタビン1回1,000若しくは1,250mg/m2、シスプラチン1回75mg/m2を3週間間隔で最大3サイクル点滴静注、又はニボルマブ(遺伝子組換え)1回360mg、パクリタキセル1回175若しくは200mg/m2、カルボプラチン1回AUC5若しくは6(mg/mL・min)を3週間間隔で最大3サイクル点滴静注した。ゲムシタビンは各サイクル1日目及び8日目に点滴静注した。
非扁平上皮癌に対しては、ニボルマブ(遺伝子組換え)1回360mg、ペメトレキセド1回500mg/m2、シスプラチン1回75mg/m2を3週間間隔で最大3サイクル点滴静注、又はニボルマブ(遺伝子組換え)1回360mg、パクリタキセル1回175若しくは200mg/m2、カルボプラチン1回AUC5若しくは6(mg/mL・min)を3週間間隔で最大3サイクル点滴静注した。
なお、シスプラチンに対する忍容性がないと判断された場合には、シスプラチンをカルボプラチン1回AUC5若しくは6(mg/mL・min)に変更可能とされた。
併用投与時においては、ニボルマブ(遺伝子組換え)を最初に投与し、化学療法はニボルマブ(遺伝子組換え)の投与終了から約30分の間隔をおいて投与を開始した。
非扁平上皮癌に対しては、ニボルマブ(遺伝子組換え)1回360mg、ペメトレキセド1回500mg/m2、シスプラチン1回75mg/m2を3週間間隔で最大3サイクル点滴静注、又はニボルマブ(遺伝子組換え)1回360mg、パクリタキセル1回175若しくは200mg/m2、カルボプラチン1回AUC5若しくは6(mg/mL・min)を3週間間隔で最大3サイクル点滴静注した。
なお、シスプラチンに対する忍容性がないと判断された場合には、シスプラチンをカルボプラチン1回AUC5若しくは6(mg/mL・min)に変更可能とされた。
併用投与時においては、ニボルマブ(遺伝子組換え)を最初に投与し、化学療法はニボルマブ(遺伝子組換え)の投与終了から約30分の間隔をおいて投与を開始した。
17.1.7 国際共同第III相試験(KEYNOTE-671試験)
臨床病期II期、IIIA期又はIIIB期の周術期の非小細胞肺癌患者注22)797例(日本人82例を含む)を対象に、術前補助療法としてのペムブロリズマブと化学療法との併用療法、及び術後補助療法としてのペムブロリズマブ単独療法注23)の有効性及び安全性が、術前補助療法としてのプラセボと化学療法との併用療法、及び術後補助療法としてのプラセボ投与注24)を対照とした二重盲検試験で検討された10)。
主要評価項目は全生存期間(OS)及び無イベント生存期間(EFS)とされ、術前補助療法としてのペムブロリズマブと化学療法との併用療法、及び術後補助療法としてのペムブロリズマブ単独療法は、術前補助療法としてのプラセボと化学療法との併用療法、及び術後補助療法としてのプラセボ投与と比較してOS及びEFSを有意に延長した(表6、図6及び図7)。
ペムブロリズマブ、ペメトレキセド及びシスプラチンが併用投与された患者における安全性解析対象例222例中216例(97.3%)(日本人22例中22例を含む)に副作用が認められた。主な副作用は、悪心127例(57.2%)、好中球数減少87例(39.2%)、疲労66例(29.7%)、貧血63例(28.4%)、便秘63例(28.4%)、白血球数減少58例(26.1%)及び食欲減退54例(24.3%)であった(2023年7月10日データカットオフ)。[5.4、7.6参照]
主要評価項目は全生存期間(OS)及び無イベント生存期間(EFS)とされ、術前補助療法としてのペムブロリズマブと化学療法との併用療法、及び術後補助療法としてのペムブロリズマブ単独療法は、術前補助療法としてのプラセボと化学療法との併用療法、及び術後補助療法としてのプラセボ投与と比較してOS及びEFSを有意に延長した(表6、図6及び図7)。
ペムブロリズマブ、ペメトレキセド及びシスプラチンが併用投与された患者における安全性解析対象例222例中216例(97.3%)(日本人22例中22例を含む)に副作用が認められた。主な副作用は、悪心127例(57.2%)、好中球数減少87例(39.2%)、疲労66例(29.7%)、貧血63例(28.4%)、便秘63例(28.4%)、白血球数減少58例(26.1%)及び食欲減退54例(24.3%)であった(2023年7月10日データカットオフ)。[5.4、7.6参照]
注22)臨床病期はAmerican Joint Committee on Cancer(AJCC)/Union for International Cancer Control(UICC)病期分類(第8版)に基づく。なお、IIIB期はT3N2M0、T4N2M0に該当する患者が対象とされた。EGFR遺伝子変異陽性又はALK融合遺伝子陽性の患者を含む。
| 術前補助療法/術後補助療法 | ペムブロリズマブと化学療法との併用療法/ペムブロリズマブ注23)(397例) | プラセボと化学療法との併用療法/プラセボ注24)(400例) | |
| OS注25) | 中央値[月] (95%信頼区間) | NE (NE,NE) | 52.4 (45.7,NE) |
| ハザード比注26) (95%信頼区間) P値注27) | 0.72 (0.56,0.93) 0.00517 | ||
| EFS注28) | 中央値[月] (95%信頼区間) | NE (34.1,NE) | 17.0 (14.3,22.0) |
| ハザード比注26) (95%信頼区間) P値注27) | 0.58 (0.46,0.72) <0.00001 | ||
注23)術前補助療法としてペムブロリズマブ200mg 3週間間隔投与(Q3W)(各コースの1日目に投与)と以下の化学療法を最大4コース併用し、術後補助療法としてペムブロリズマブ200mg Q3W(各コースの1日目に投与)を13コース投与した[扁平上皮非小細胞肺癌:ゲムシタビン1,000mg/m2(1コース21日間、各コースの1、8日目に投与)、シスプラチン75mg/m2(1コース21日間、各コースの1日目に投与)を投与。非扁平上皮非小細胞肺癌:ペメトレキセド500mg/m2、シスプラチン75mg/m2をQ3Wで各コースの1日目に投与]。
注24)術前補助療法としてプラセボQ3W(各コースの1日目に投与)と注23)と同一の化学療法を最大4コース併用し、術後補助療法としてプラセボQ3W(各コースの1日目に投与)を13コース投与した。
注25)中間解析時のデータ:2023年7月10日カットオフ
注26)層別Cox比例ハザードモデルによるプラセボと化学療法との併用療法/プラセボとの比較
注27)層別ログランク検定
注28)中間解析時のデータ:2022年7月29日カットオフ(EFSは治験担当医師による評価)
NE:Not Estimated
図6)OSのKaplan-Meier曲線(KEYNOTE-671試験)
図7)EFSのKaplan-Meier曲線(KEYNOTE-671試験)
18. 薬効薬理
18.1 作用機序
18.2 抗腫瘍効果
19. 有効成分に関する理化学的知見
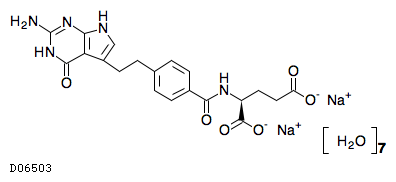
19.1. ペメトレキセドナトリウム水和物
| 一般的名称 | ペメトレキセドナトリウム水和物 |
|---|---|
| 一般的名称(欧名) | Pemetrexed Sodium Hydrate |
| 化学名 | Disodium N-{4-[2-(2-amino-4-oxo-4,7-dihydro-1H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl}-L-glutamate heptahydrate |
| 分子式 | C20H19N5Na2O6・7H2O |
| 分子量 | 597.48 |
| 物理化学的性状 | 白色の粉末又は塊である。 水に溶けやすく、メタノールにやや溶けやすく、エタノール(99.5)に極めて溶けにくい。 |
| KEGG DRUG |
|
22. 包装
<アリムタ注射用100mg>
100mg[1バイアル]
<アリムタ注射用500mg>
500mg[1バイアル]
23. 主要文献
- Nakagawa K,et al., Br.J.Cancer., 95, 677-682, (2006) »PubMed
- Chay S.H,et al., Proc.Amer.Assoc.Cancer Res., 39, 524-525, (1998)
- 社内資料:ヒト血漿におけるin vitro蛋白結合率(2007年1月4日承認、CTD 2.7.2.3.1.2)
- 社内資料:クリアランスと腎機能との関連(2007年1月4日承認、CTD 2.7.2.3.4.1.4)
- 社内資料:薬物動態学的相互作用(2007年1月4日承認、CTD 2.7.2.3.5.2.2、2.7.2.3.3.1)
- Vogelzang N.J,et al., J.Clin.Oncol., 21, 2636-2644, (2003) »PubMed
- Scagliotti G.V,et al., J.Clin.Oncol., 26, 3543-3551, (2008) »PubMed
- Scagliotti G.V,et al., The Oncologist., 14, 253-263, (2009)
- Hanna N,et al., J.Clin.Oncol., 22, 1589-1597, (2004) »PubMed
- Heather W,et al., N.Engl.J.Med., 389, 491-503, (2023)
- Habeck L.L,et al., Mol.Pharmacol., 48, 326-333, (1995) »PubMed
- Zhao R,et al., Clin.Cancer Res., 6, 3687-3695, (2000) »PubMed
- Shih C,et al., Cancer Res., 57, 1116-1123, (1997) »PubMed
- Britten C.D,et al., Cancer Chemother.Pharmacol., 44, 105-110, (1999) »PubMed
- Chan D.C,et al., Proc.Amer.Assoc.Cancer Res., 47, 1278, (2006)
24. 文献請求先及び問い合わせ先
文献請求先
日本イーライリリー株式会社
医薬情報問合せ窓口
〒651-0086
神戸市中央区磯上通5丁目1番28号
電話:0120-360-605(医療関係者向け)
製品情報問い合わせ先
日本イーライリリー株式会社
医薬情報問合せ窓口
〒651-0086
神戸市中央区磯上通5丁目1番28号
電話:0120-360-605(医療関係者向け)
26. 製造販売業者等
26.1 製造販売元
日本イーライリリー株式会社
神戸市中央区磯上通5丁目1番28号