医薬品情報
| 総称名 | メサペイン |
|---|---|
| 一般名 | メサドン塩酸塩 |
| 欧文一般名 | Methadone Hydrochloride |
| 製剤名 | メサドン塩酸塩錠 |
| 薬効分類名 | がん疼痛治療剤 |
| 薬効分類番号 | 8219 |
| ATCコード | N07BC02 |
| KEGG DRUG |
D02102
メサドン塩酸塩
|
| JAPIC | 添付文書(PDF) |
添付文書情報2024年3月 改訂(第1版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| メサペイン錠5mg | METHAPAIN Tablets 5mg | 帝國製薬 | 8219002F1024 | 184.8円/錠 | 劇薬, 麻薬, 処方箋医薬品注) |
| メサペイン錠10mg | METHAPAIN Tablets 10mg | 帝國製薬 | 8219002F2020 | 351.2円/錠 | 劇薬, 麻薬, 処方箋医薬品注) |
1. 警告
2. 禁忌
次の患者には投与しないこと
4. 効能または効果
他の強オピオイド鎮痛剤で治療困難な下記疾患における鎮痛
中等度から高度の疼痛を伴う各種癌
5. 効能または効果に関連する注意
本剤は、他の強オピオイド鎮痛剤の投与では十分な鎮痛効果が得られない患者で、かつオピオイド鎮痛剤の継続的な投与を必要とするがん疼痛の管理にのみ使用すること。[14.1.1参照]
6. 用法及び用量
本剤は、他の強オピオイド鎮痛剤から切り替えて使用する。
通常、成人に対し初回投与量は本剤投与前に使用していた強オピオイド鎮痛剤の用法・用量を勘案して、メサドン塩酸塩として1回5〜15mgを1日3回経口投与する。
その後の投与量は患者の症状や状態により適宜増減する。
通常、成人に対し初回投与量は本剤投与前に使用していた強オピオイド鎮痛剤の用法・用量を勘案して、メサドン塩酸塩として1回5〜15mgを1日3回経口投与する。
その後の投与量は患者の症状や状態により適宜増減する。
7. 用法及び用量に関連する注意
7.1 初回投与量
7.1.1 本剤の薬物動態は個人差が大きく、他のオピオイド鎮痛剤との交差耐性が不完全であるため、本剤と他のオピオイド鎮痛剤の等鎮痛比は確立していない。[1.3参照]
7.1.2 下記換算表は、初回投与量を選択する際の目安であり、換算比は本剤投与前に使用していたオピオイド鎮痛剤の投与量により大幅に異なる。患者の症状や状態、オピオイド耐性の程度、併用薬剤を考慮して適切な用量を選択し、過量投与にならないよう注意すること。[1.3参照]
7.1.3 経口モルヒネ量60mg/日未満のオピオイド鎮痛剤からの切り替えは推奨されない。
| メサドン塩酸塩(mg/日) | 15mg/日(5mg/回×3回) | 30mg/日(10mg/回×3回) | 45mg/日(15mg/回×3回) |
| モルヒネ経口剤(mg/日) | 60≦〜≦160 | 160<〜≦390 | 390< |
7.2 初回投与時
7.2.1 本剤投与後少なくとも7日間は増量を行わないこと。本剤の血中濃度が定常状態に達するまでに時間を要することから、7日未満の増量は過量投与となる可能性がある。[1.3、7.4.1、16.1.2参照]
7.2.2 フェンタニル貼付剤から本剤へ変更する場合には、フェンタニル貼付剤剥離後にフェンタニルの血中濃度が50%に減少するまで17時間以上かかることから、剥離直後の本剤の使用は避け、本剤の使用を開始するまでに、フェンタニルの血中濃度が適切な濃度に低下するまでの時間をあけるとともに、本剤の低用量から投与することを考慮すること。
7.3 疼痛増強時
本剤服用中に疼痛が増強した場合や鎮痛効果が得られている患者で突発性の疼痛が発現した場合は、直ちに速効性のオピオイド鎮痛剤の臨時追加投与(レスキュー薬の投与)を行い鎮痛を図ること。
7.4 増量
7.4.2 鎮痛効果が得られるまで患者毎に用量調整を行うこと。鎮痛効果が得られない場合は、1日あたり本剤1日投与量の50%、1回あたり5mgを上限に増量すること。
7.4.3 本剤を増量する場合には、副作用に十分注意すること。[1.3参照]
7.5 減量
連用中における急激な減量は、退薬症候があらわれることがあるので行わないこと。副作用等により減量する場合は、患者の状態を観察しながら慎重に行うこと。
7.6 投与の中止
本剤の投与を中止する場合には、退薬症候の発現を防ぐために徐々に減量すること。副作用等により直ちに投与を中止する場合は、退薬症候の発現に注意すること。
8. 重要な基本的注意
8.1 本剤の投与開始にあたっては、主な副作用、相互作用、投与時の注意点等を患者等に対して十分に説明し、理解を得た上で投与を開始すること。特に不整脈、呼吸抑制等の症状が認められた場合には、速やかに主治医に連絡するよう指導すること。[1.2、1.3、2.1、8.2-8.4、9.1.3-9.1.5、11.1.3、11.1.4、14.1.2参照]
8.2 高用量の強オピオイド鎮痛剤からの切り替え、呼吸抑制を起こしやすい患者等では、入院又はそれに準じる管理の下で本剤の投与開始及び用量調節を行うなど、重篤な副作用発現に関する観察を十分に行うこと。[1.2、2.1、8.1、8.4、9.1.5、11.1.3参照]
8.3 QT延長があらわれることがあるので、本剤投与開始前及び本剤投与中は定期的に心電図検査及び電解質検査を行い、患者の状態を十分に観察すること。特に、本剤1日投与量が100mgを超える前及びその1週間後、QT延長を起こしやすい患者では、本剤の投与量が安定した時点で心電図検査を行うことが望ましい。異常が認められた場合には、必要に応じて休薬、減量又は中止し、適切な処置を行うこと。[1.2、8.1、9.1.3、9.1.4、11.1.4、13.2.3参照]
8.4 重篤な呼吸抑制が認められた場合には、投与を中止し、呼吸管理を行うこと。呼吸抑制に対しては麻薬拮抗剤(ナロキソン、レバロルファン等)が有効であるが、麻薬拮抗剤の作用持続時間は本剤より短いので、観察を十分に行い麻薬拮抗剤の繰り返し投与を考慮すること。[1.2、2.1、8.1、8.2、9.1.5、11.1.3、13.2.2参照]
8.5 本剤を投与する場合には、便秘に対する対策として緩下剤、嘔気・嘔吐に対する対策として制吐剤の併用を、また、鎮痛効果が得られている患者で通常とは異なる強い眠気がある場合には、過量投与の可能性を念頭において本剤の減量を考慮するなど、本剤投与時の副作用に十分注意すること。
8.7 重篤な副作用が発現した患者については、本剤の血中動態を考慮し、投与中止時から少なくとも48時間後まで観察を継続すること。
8.8 眠気、めまいが起こることがあるので、本剤投与中の患者には自動車の運転等危険を伴う機械の操作に従事させないように注意すること。
8.9 本剤は種々の薬剤との相互作用が報告されていることから、併用薬剤に十分注意して投与すること。[10.参照]
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 細菌性下痢のある患者
治療上やむを得ないと判断される場合を除き、投与しないこと。治療期間の延長を来すおそれがある。
9.1.2 心機能障害又は低血圧のある患者
循環不全を増強するおそれがある。[11.1.4参照]
9.1.3 QT延長のある患者
9.1.4 QT延長を起こしやすい患者
9.1.5 呼吸機能障害のある患者
9.1.6 脳に器質的障害のある患者
呼吸抑制や頭蓋内圧の上昇を起こすおそれがある。
9.1.7 ショック状態にある患者
循環不全や呼吸抑制を増強するおそれがある。
9.1.8 代謝性アシドーシスのある患者
呼吸抑制を起こすおそれがある。
9.1.9 てんかん等の痙攣性疾患又はこれらの既往歴のある患者
痙攣を起こすおそれがある。
9.1.10 甲状腺機能低下症(粘液水腫等)、副腎皮質機能低下症(アジソン病等)又は衰弱者
呼吸抑制作用に対し、感受性が高くなっている。
9.1.11 薬物依存の既往歴のある患者
9.1.12 前立腺肥大による排尿障害、尿道狭窄、尿路手術術後の患者
排尿障害を増悪することがある。
9.1.13 器質的幽門狭窄、重篤な炎症性腸疾患又は最近消化管手術を行った患者
消化管運動を抑制する。
9.1.14 胆嚢障害、胆石症又は膵炎の患者
オッジ筋を収縮させ症状が増悪することがある。
9.2 腎機能障害患者
排泄が遅延し副作用があらわれるおそれがある。
9.3 肝機能障害患者
代謝が遅延し副作用があらわれるおそれがある。
9.5 妊婦
9.6 授乳婦
授乳を避けさせること。ヒト母乳中へ移行し、母親の経口投与量が10〜80mg/日のとき、メサドンの乳汁中濃度は0.05〜0.57μg/mLになることが報告されている6)。
9.7 小児等
小児等を対象とした臨床試験は実施していない。
9.8 高齢者
低用量から投与を開始するなど患者の状態を観察しながら、慎重に投与すること。一般に生理機能が低下しており、特に呼吸抑制の感受性が高い。
10. 相互作用
相互作用序文
薬物代謝酵素用語
CYP3A4
薬物代謝酵素用語
CYP2B6
薬物代謝酵素用語
CYP2C8
薬物代謝酵素用語
CYP2C9
薬物代謝酵素用語
CYP2C19
薬物代謝酵素用語
CYP2D6
薬物代謝酵素用語
P糖蛋白
10.1 併用禁忌
| ナルメフェン塩酸塩水和物 (セリンクロ) [2.7参照] | ナルメフェン塩酸塩水和物により本剤の鎮痛作用を減弱させるため、効果を得るために必要な用量が通常用量より多くなるおそれがある。 また、退薬症候を起こすおそれがある。 | ナルメフェン塩酸塩水和物はμ受容体のアンタゴニストであり、μ受容体のアゴニストである本剤に対して、競合的に阻害する。 |
10.2 併用注意
| QT延長を起こすことが知られている薬剤 スニチニブ、ダサチニブ、マプロチリン等 抗不整脈剤 ジソピラミド、プロカインアミド、アミオダロン、ソタロール等 抗精神病剤 | 不整脈を誘発するおそれがある。 | 相加的にQT延長作用を増強させる。 |
| 低カリウム血症を起こす薬剤 利尿剤 副腎皮質ステロイド剤等 | 低カリウム血症による不整脈を誘発するおそれがある。 | カリウム値の低下により心臓の不応期が延長され、さらに本剤の投与により新たな不整脈を誘発することによる。 |
| 三環系抗うつ剤 イミプラミン、アミトリプチリン等 | 不整脈を誘発するおそれがある。 | 相加的にQT延長作用を増強させる。 |
| 三環系抗うつ剤 イミプラミン、アミトリプチリン等 | 呼吸抑制、低血圧及び顕著な鎮静又は昏睡が起こるおそれがあるので、減量するなど慎重に投与すること。 | 相加的に中枢神経抑制作用を増強させる。 |
| 中枢神経抑制剤 ベンゾジアゼピン誘導体、フェノチアジン誘導体、バルビツール酸誘導体等 アルコール 吸入麻酔剤 MAO阻害剤 オピオイド鎮痛剤 | 呼吸抑制、低血圧及び顕著な鎮静又は昏睡が起こるおそれがあるので、減量するなど慎重に投与すること。 | 相加的に中枢神経抑制作用を増強させる。 |
| 選択的セロトニン再取り込み阻害剤 セルトラリン塩酸塩、フルボキサミンマレイン酸塩等 | 本剤の血中濃度が増加したとの報告がある8)。 | 機序不明 |
| 尿アルカリ化を起こす薬剤 炭酸水素ナトリウム等 | 本剤の血中濃度が増加したとの報告がある9)。 | 尿のアルカリ化により本剤の尿中排泄率が低下するため。 |
| 抗真菌剤 ケトコナゾール注)、ボリコナゾール等 マクロライド系抗菌剤 エリスロマイシン等 | 本剤の血中濃度が増加するおそれがある。 | これらの薬剤が本剤の肝薬物代謝酵素(CYP3A4)を阻害することによる。 |
| 肝代謝酵素誘導作用を有する薬剤 リファンピシン、フェニトイン、フェノバルビタール、カルバマゼピン | 本剤の血中濃度が低下したとの報告がある10)。 | これらの薬剤が本剤の肝薬物代謝酵素(CYP3A4等)を誘導することによる。 |
| セイヨウオトギリソウ(St.John's Wort、セント・ジョーンズ・ワート)含有食品 | 本剤の血中濃度が低下するおそれがある。 | セイヨウオトギリソウが本剤の肝薬物代謝酵素(CYP3A4)を誘導することによる。 |
| アバカビル硫酸塩、エファビレンツ、ネビラピン、リルピビリン塩酸塩、ロピナビル・リトナビル配合剤 | 本剤の血中濃度が低下したとの報告がある11)。 | 機序不明 |
| ジドブジン (アジドチミジン) | ジドブジンの血中濃度が増加したとの報告がある12)。 | 機序不明 |
| 抗コリン作用を有する薬剤 | 麻痺性イレウスに至る重篤な便秘又は尿貯留が起こるおそれがある。 | 相加的に抗コリン作用を増強させる。 |
| ブプレノルフィン、ペンタゾシン | 本剤の鎮痛作用を減弱させることがある。また、退薬症候を起こすおそれがある。 | これらの薬剤は本剤の作用するμ受容体の部分アゴニストである。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 ショック、アナフィラキシー(いずれも頻度不明)
顔面蒼白、血圧低下、呼吸困難、頻脈、全身発赤、血管浮腫、蕁麻疹等の症状があらわれた場合には投与を中止し、適切な処置を行うこと。
11.1.2 依存性(頻度不明)
11.1.3 呼吸停止、呼吸抑制(いずれも頻度不明)
11.1.4 心停止、心室細動、心室頻拍(Torsade de pointesを含む)、心不全、期外収縮(いずれも頻度不明)、QT延長(15.4%)[1.2、8.1、8.3、9.1.2-9.1.4、13.2.3参照]
11.1.5 錯乱(頻度不明)、せん妄(7.7%)
11.1.6 肺水腫、無気肺、気管支痙攣、喉頭浮腫(いずれも頻度不明)
11.1.7 腸閉塞(3.8%)、麻痺性イレウス、中毒性巨大結腸(いずれも頻度不明)[2.3参照]
11.1.8 肝機能障害(頻度不明)
AST(GOT)、ALT(GPT)、Al-P等の著しい上昇を伴う肝機能障害があらわれることがある。
11.2 その他の副作用
| 10%以上 | 10%未満 | 頻度不明 | |
| 循環器 | 不整脈、二段脈、徐脈、頻脈、T波逆転、血圧変動、失神、心筋症、動悸 | ||
| 精神神経系 | 眠気・傾眠 | 振戦 | 不眠、めまい、ふらふら感、幻覚、健忘、失見当識、激越、不安、鎮静、気分不快、多幸感、感覚異常、痙攣発作、頭痛、発汗、ミオクローヌス |
| 消化器 | 悪心、嘔吐、便秘 | 下痢 | 腹痛、口渇、味覚異常、食欲不振、舌炎、胆管痙攣 |
| 過敏症 | 発疹 | そう痒症 | |
| 血液 | 血小板減少症 | ||
| 泌尿器 | 排尿障害、尿閉 | ||
| 感覚器 | 視覚障害(霧視、複視等) | ||
| その他 | 血管拡張(顔面潮紅、熱感)、潮紅、浮腫、呼吸困難、無力症、脱力、倦怠感、低カリウム血症、低マグネシウム血症、静脈炎、体重増加、無月経、性欲減退、性能力減退 |
13. 過量投与
13.1 徴候・症状
呼吸抑制、意識不明、痙攣、錯乱、血圧低下、重篤な脱力感、重篤なめまい、嗜眠、心拍数の減少、QT延長、心室頻拍(Torsade de pointesを含む)、神経過敏、不安、縮瞳、皮膚冷感、無呼吸、循環虚脱等を起こすことがある。[1.3参照]
13.2 処置
14. 適用上の注意
14.1 薬剤交付時の注意
14.1.1 強オピオイド鎮痛剤が投与されていた患者であることを確認した上で本剤を交付すること。[5.参照]
14.1.2 本剤の投与開始にあたっては、患者等に対して、主な副作用、相互作用、具体的な服用方法、服用時の注意点、保管方法等を患者向けの説明書を用いるなどの方法によって十分に説明すること。[8.1、8.10参照]
14.1.3 患者等に対して、本剤の目的以外への使用あるいは他人への譲渡をしないよう指導するとともに、本剤を子供の手の届かないところに保管するよう指導すること。[8.10参照]
14.1.4 PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔を起こして縦隔洞炎等の重篤な合併症を併発することがある。
14.1.5 本剤が不要となった場合には、病院又は薬局へ返納するなどの処置について適切に指導すること。[8.10参照]
15. その他の注意
15.2 非臨床試験に基づく情報
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
メサドン塩酸塩錠5mg、10mgを健康成人男性(外国在住日本人、6例)に単回経口投与したとき、最高血漿中濃度(Cmax)及び血漿中濃度曲線下面積(AUC0-144hr)は用量に比例して増加した。なお、メサドン塩酸塩錠5mg又は10mgを単回投与したときのメサドン及び主代謝物2-ethylidene-1,5-dimethyl-3,3-diphenylpyrrolidine(EDDP)の薬物動態パラメータは表1のとおりであった18)。
図1 単回経口投与時の血漿中メサドン濃度推移
| n | AUC0-144hr(ng・hr/mL) | Cmax(ng/mL) | tmax(hr) | t1/2(hr) | ||
| メサドン塩酸塩錠5mg | メサドン | 6 | 572.0±161.0 | 15.6±4.1 | 4.9±2.1 | 37.2±4.6 |
| 主代謝物(EDDP) | 51.4±14.0 | 1.5±0.5 | 5.9±3.6 | 40.3±12.3 | ||
| メサドン塩酸塩錠10mg | メサドン | 6 | 1026.5±239.3 | 30.8±4.6 | 3.3±2.4 | 38.3±4.9 |
| 主代謝物(EDDP) | 94.2±16.0 | 3.4±0.6 | 2.2±0.6 | 28.3±9.7 | ||
16.1.2 反復投与時
16.3 分布
16.3.1 血漿蛋白結合率
平衡透析法で測定した血漿蛋白質との結合率は、89.4%であり、主にα1-酸性糖蛋白に結合する19)(健康成人)(外国人データ)。
16.4 代謝
16.4.1 メサドン塩酸塩を麻薬中毒患者に経口投与したとき、メサドンはN-脱メチル化、環化により主に肝臓で代謝される。主な代謝物は不活性な2-ethylidene-1,5-dimethyl-3,3-diphenylpyrrolidine(EDDP)である20)(外国人データ)。
16.5 排泄
14C標識体メサドンを麻薬中毒患者(男性)に経口投与したとき、放射能の尿中及び糞中排泄率はそれぞれ24〜79%及び18〜42%であった23)(外国人データ)。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国内第II相試験(オピオイド鎮痛剤を使用しているがん疼痛患者を対象とした切り替え試験)
他のオピオイド鎮痛剤(モルヒネ、オキシコドン、フェンタニル)により十分な疼痛管理が得られないがん疼痛患者(目標症例数:20例)を対象とした本剤への切り替え試験において、疼痛コントロールの達成率(至適用量に到達注1)した患者)は85.0%(17例/20例)であった。また、切り替え前後の疼痛強度(NRS注2))変化量の平均値とその95%信頼区間は-1.9[-2.7、-0.99]であった24)。
注1)同一用量で6日間以上投与した翌日に以下の至適用量到達の判定基準をすべて満たす場合に、切り替えにより疼痛コントロールを達成したと判定した
<至適用量到達の判定基準>
1)評価日前日のレスキュー使用回数が1日2回以下であって、かつNRS注2)が切り替え前の値以下である
2)忍容できない副作用及び過量投与の兆候がない
3)その他、安全性・有効性観点から、投与量に問題ありと考えられる要因がない
注2)Numerical Rating Scale(「痛くない(0)」から「想像できる最大の痛み(10)」までの11段階の評価スケール)
17.1.2 国内臨床試験全期(第II相試験+長期投与試験)
本剤の承認時までに実施した国内臨床試験において、総症例26例中20例(76.9%)に43件の副作用(臨床検査値の異常変動を含む)が認められた。主な副作用は、傾眠13例(50.0%)、悪心6例(23.1%)、嘔吐5例(19.2%)、心電図QT延長4例(15.4%)、便秘4例(15.4%)及びせん妄2例(7.7%)等であった25)。
17.2 製造販売後調査等
17.2.1 国内使用成績調査
使用成績調査において、安全性解析対象症例816例中360例(44.1%)に605件の副作用(臨床検査値の異常変動を含む)が認められた。主な副作用は、傾眠205例(25.1%)、悪心63例(7.7%)、せん妄及び便秘各45例(各5.5%)、嘔吐28例(3.4%)等であった。心電図QT延長は23例(2.8%)、Torsade de pointesは1例(0.1%)認められた。また呼吸抑制は10例(1.2%)、薬剤離脱症候群は1例(0.1%)認められた26)(再審査終了時)。
18. 薬効薬理
18.1 作用機序
モルヒネと同様にμオピオイド受容体を介して鎮痛作用を示すものと考えられており、メサドンはμオピオイド受容体に対して高い親和性が認められた27)。
18.2 鎮痛作用
ラットを用い熱刺激法(温湯法)により鎮痛効果を検討した結果、その効果はモルヒネよりやや強くオキシコドンと同程度であった27)。
19. 有効成分に関する理化学的知見
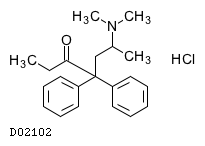
19.1. メサドン塩酸塩
| 一般的名称 | メサドン塩酸塩 |
|---|---|
| 一般的名称(欧名) | Methadone Hydrochloride |
| 化学名 | (6RS)-6-(Dimethylamino)-4,4-diphenylheptan-3-one monohydrochloride |
| 分子式 | C21H27NO・HCl |
| 分子量 | 345.91 |
| 物理化学的性状 | 無色の結晶又は白色の結晶性の粉末である。 エタノール(95)に溶けやすく、水にやや溶けやすく、ジエチルエーテル及びグリセリンにほとんど溶けない。 |
| 理化学知見その他 | 本品の水溶液(1→100)のpHは4.5〜6.5である。 |
| KEGG DRUG |
|
20. 取扱い上の注意
アルミ袋開封後は直射日光、高温、多湿を避けて保存すること。
21. 承認条件
がん性疼痛の治療に精通した医師によってのみ処方・使用されるとともに、本剤のリスク等についても十分に管理・説明できる医師・医療機関・管理薬剤師のいる薬局のもとでのみ用いられ、それら薬局においては調剤前に当該医師・医療機関を確認した上で調剤がなされるよう、製造販売にあたって必要な措置を講じること。
22. 包装
<メサペイン錠5mg>
40錠(10錠×4)PTP
100錠(10錠×10)PTP
<メサペイン錠10mg>
40錠(10錠×4)PTP
100錠(10錠×10)PTP
23. 主要文献
- Buchenauer D,et al., J Pharmacol Exp Ther., 189 (1), 66-71, (1974) »PubMed
- Hutchings DE,et al., J Pharmacol Exp Ther., 197 (1), 171-179, (1976) »PubMed
- Bui QQ,et al., Drug Chem Toxicol., 6 (1), 41-70, (1983) »PubMed
- Geber WF,et al., Am J Obstet Gynecol., 123 (7), 705-713, (1975) »PubMed
- Dashe JS,et al., Obstet Gynecol., 100 (6), 1244-1249, (2002) »PubMed
- Blinick G,et al., Am J Obstet Gynecol., 121 (5), 617-621, (1975) »PubMed
- Tolson AH,et al., Drug Metab Dispos., 37 (9), 1887-1894, (2009) »PubMed
- Eap CB,et al., J Clin Psychopharmacol., 17 (2), 113-117, (1997) »PubMed
- Nilsson MI,et al., Eur J Clin Pharmacol., 22 (4), 337-342, (1982) »PubMed
- Tong TG,et al., Ann Intern Med., 94 (3), 349-351, (1981) »PubMed
- Clarke SM,et al., Br J Clin Pharmacol., 51 (3), 213-217, (2001) »PubMed
- McCance-Katz EF,et al., J Acquir Immune Defic Syndr Hum Retrovirol., 18 (5), 435-443, (1998) »PubMed
- Badr FM,et al., Mutat Res., 68 (3), 235-249, (1979) »PubMed
- Brusick D,et al., Drug Chem Toxicol., 4 (1), 1-18, (1981) »PubMed
- Smith DJ,et al., Nature., 253 (5488), 202-203, (1975) »PubMed
- Murphy MR., Pharmacol Biochem Behav., 14 (4), 561-567, (1981) »PubMed
- Rosenkrantz H,et al., Fundam Appl Toxicol., 11 (4), 640-651, (1988) »PubMed
- 社内資料:健康成人における薬物動態に関する資料
- Romach MK,et al., Clin Pharmacol Ther., 29 (2), 211-217, (1981) »PubMed
- Pohland A.et al., J Med Chem., 14 (3), 194-197, (1971)
- Wang JS,et al., Drug Metab Dispos., 31 (6), 742-747, (2003) »PubMed
- Totah RA,et al., Anesthesiology., 108 (3), 363-374, (2008) »PubMed
- Anggard E,et al., Clin Pharmacol Ther., 17 (3), 258-266, (1975) »PubMed
- 社内資料:がん疼痛患者における切り替え臨床試験に関する資料
- 社内資料:有害事象に関する資料(第II相試験+長期投与試験)
- 社内資料:使用成績調査(全例調査)最終報告
- Peckham EM,et al., J Pharmacol Exp Ther., 316 (3), 1195-1201, (2006) »PubMed
24. 文献請求先及び問い合わせ先
文献請求先
テルモ株式会社
テルモ・コールセンター
〒259-0151
神奈川県足柄上郡中井町井ノ口1500
電話:0120-12-8195
製品情報問い合わせ先
テルモ株式会社
テルモ・コールセンター
〒259-0151
神奈川県足柄上郡中井町井ノ口1500
電話:0120-12-8195
25. 保険給付上の注意
本剤は厚生労働省告示第107号(平成18年3月6日付)に基づき、投薬は1回14日分を限度とされています。
26. 製造販売業者等
26.1 製造販売元
帝國製薬株式会社
香川県東かがわ市三本松567番地
26.2 販売元
テルモ株式会社
東京都渋谷区幡ヶ谷2-44-1