医薬品情報
| 総称名 | プラミペキソール塩酸塩 |
|---|---|
| 一般名 | プラミペキソール塩酸塩水和物 |
| 欧文一般名 | Pramipexole Hydrochloride Hydrate |
| 製剤名 | プラミペキソール塩酸塩水和物錠 |
| 薬効分類名 | ドパミン作動性パーキンソン病治療剤 レストレスレッグス症候群治療剤 |
| 薬効分類番号 | 1169 1190 |
| ATCコード | N04BC05 |
| KEGG DRUG |
D00559
プラミペキソール塩酸塩水和物
|
| KEGG DGROUP |
DG01967
抗パーキンソン病薬
|
| JAPIC | 添付文書(PDF) |
添付文書情報2024年6月 改訂(第2版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| プラミペキソール塩酸塩錠0.125mg「FFP」 (後発品) | Pramipexole Hydrochloride Tablets 0.125mg「FFP」 | 共創未来ファーマ | 1169012F1065 | 8.9円/錠 | 劇薬, 処方箋医薬品注) |
| プラミペキソール塩酸塩錠0.5mg「FFP」 (後発品) | Pramipexole Hydrochloride Tablets 0.5mg「FFP」 | 共創未来ファーマ | 1169012F2061 | 33.1円/錠 | 劇薬, 処方箋医薬品注) |
1. 警告
2. 禁忌
次の患者には投与しないこと
2.1 妊婦又は妊娠している可能性のある女性[9.5参照]
2.2 本剤の成分に対し過敏症の既往歴のある患者
4. 効能または効果
○中等度から高度の特発性レストレスレッグス症候群(下肢静止不能症候群)
5. 効能または効果に関連する注意
レストレスレッグス症候群(下肢静止不能症候群)の診断は、国際レストレスレッグス症候群研究グループの診断基準及び重症度スケールに基づき慎重に実施し、基準を満たす場合にのみ投与すること。
6. 用法及び用量
<パーキンソン病>
通常、成人にはプラミペキソール塩酸塩水和物として1日量0.25mgからはじめ、2週目に1日量を0.5mgとし、以後経過を観察しながら、1週間毎に1日量として0.5mgずつ増量し、維持量(標準1日量1.5〜4.5mg)を定める。1日量がプラミペキソール塩酸塩水和物として1.5mg未満の場合は2回に分割して朝夕食後に、1.5mg以上の場合は3回に分割して毎食後経口投与する。なお、年齢、症状により適宜増減ができるが、1日量は4.5mgを超えないこと。
<中等度から高度の特発性レストレスレッグス症候群(下肢静止不能症候群)>
通常、成人にはプラミペキソール塩酸塩水和物として0.25mgを1日1回就寝2〜3時間前に経口投与する。投与は1日0.125mgより開始し、症状に応じて1日0.75mgを超えない範囲で適宜増減するが、増量は1週間以上の間隔をあけて行うこと。
7. 用法及び用量に関連する注意
<パーキンソン病>
7.1 本剤の投与は、少量から開始し、幻覚等の精神症状、消化器症状、血圧等の観察を十分に行い、慎重に維持量(標準1日量1.5〜4.5mg)まで増量すること。[8.2、9.1.1、9.1.3、11.1.2参照]
7.2 腎機能障害患者に対する投与法
次のような投与法を目安に投与回数を調節し腎機能に注意しながら慎重に漸増すること。なお、腎機能障害患者に対する最大1日量及び最大1回量は下表のとおりとする。[9.2.1、9.2.2、9.8.2、16.6.1参照]
| クレアチニンクリアランス(mL/min) | 投与法 | 初回1日投与量 | 最大1日量 |
| クレアチニンクリアランス≧50 | 1日量として1.5mg未満:1日2回投与 | 0.125mg×2回 | 4.5mg (1.5mg×3回) |
| 1日量として1.5mg以上:1日3回投与 | |||
| 50>クレアチニンクリアランス≧20 | 1日2回投与 | 0.125mg×2回 | 2.25mg (1.125mg×2回) |
| 20>クレアチニンクリアランス | 1日1回投与 | 0.125mg×1回 | 1.5mg (1.5mg×1回) |
<中等度から高度の特発性レストレスレッグス症候群(下肢静止不能症候群)>
8. 重要な基本的注意
<効能共通>
8.1 突発的睡眠等により自動車事故を起こした例が報告されている。突発的睡眠を起こした症例の中には、傾眠や過度の眠気のような前兆を認めなかった例あるいは投与開始後1年以上経過した後に初めて発現した例も報告されている。患者には本剤の突発的睡眠及び傾眠等についてよく説明し、自動車の運転、機械の操作、高所作業等危険を伴う作業に従事させないよう注意すること。[1.、11.1.1参照]
8.2 特に投与初期には、めまい、立ちくらみ、ふらつき等の起立性低血圧に基づく症状が見られることがある。また、これらの症状が発現した場合には、症状の程度に応じて、減量又は投与を中止するなどの適切な処置を行うこと。[7.1、9.1.2、9.1.3参照]
8.3 レボドパ又はドパミン受容体作動薬の投与により、病的賭博(個人的生活の崩壊等の社会的に不利な結果を招くにもかかわらず、持続的にギャンブルを繰り返す状態)、病的性欲亢進、強迫性購買、暴食等の衝動制御障害が報告されているので、このような症状が発現した場合には、減量又は投与を中止するなど適切な処置を行うこと。また、患者及び家族等にこのような衝動制御障害の症状について説明すること。
8.4 パーキンソン病患者において、本剤の減量、中止が必要な場合は、漸減すること。急激な減量又は中止により、悪性症候群を誘発することがある。また、ドパミン受容体作動薬の急激な減量又は中止により、薬剤離脱症候群(無感情、不安、うつ、疲労感、発汗、疼痛等の症状を特徴とする)があらわれることがある。なお、特発性レストレスレッグス症候群患者においては、パーキンソン病患者よりも用量が低いため、漸減しなくてもよい。[11.1.4参照]
<中等度から高度の特発性レストレスレッグス症候群(下肢静止不能症候群)>
8.5 本剤を含めたドパミン受容体作動薬の投与によりAugmentation(夜間の症状発現が2時間以上早まる、症状の増悪、他の四肢への症状拡大)が認められることがあるため、このような症状が認められた場合には、減量又は投与を中止するなどの適切な措置を講じること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 幻覚、妄想等の精神症状又はそれらの既往歴のある患者
9.1.2 重篤な心疾患又はそれらの既往歴のある患者
起立性低血圧等の副作用が発現しやすくなるおそれがある。[8.2参照]
9.1.3 低血圧症の患者
9.2 腎機能障害患者
9.2.1 腎機能障害のある患者
9.2.2 腎機能障害のある患者(クレアチニンクリアランスが50mL/min未満)
9.5 妊婦
妊婦又は妊娠している可能性のある女性には投与しないこと。動物(ラット)を用いた生殖発生毒性試験で、以下のことが認められている。
・受胎能及び一般生殖能試験(Seg.I)(2.5mg/kg/日投与群)で、血清プロラクチン濃度の低下に基づく妊娠率の低下
・器官形成期投与試験(Seg.II)(1.5mg/kg/日投与群)で、血清プロラクチン濃度の低下に基づく生存胎児数の減少
・周産期及び授乳期投与試験(Seg.III)(0.5mg/kg以上/日投与群)で、血清プロラクチン濃度の低下に基づく出生児体重の低下
[2.1参照]
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。ヒトにおいてプロラクチン分泌を抑制することが報告されており、乳汁分泌を抑制する可能性がある。なお、動物実験(ラット)で乳汁中へ移行することが認められている。
9.7 小児等
小児等を対象とした国内臨床試験は実施していない。
9.8 高齢者
10. 相互作用
10.2 併用注意
| カチオン輸送系を介して腎排泄される薬剤 シメチジン、アマンタジン塩酸塩 [16.7.1参照] | ジスキネジア、幻覚等の副作用が増強することがある。このような場合には、本剤を減量すること。 | カチオン輸送系を介して腎排泄される薬剤との併用により、双方あるいはいずれかの薬剤の腎尿細管分泌が減少し、腎クリアランスが低下することがある。 |
| 鎮静剤 アルコール | 作用が増強するおそれがある。 | 機序は明らかではないが、本剤との併用により作用増強の可能性が考えられる。 |
| ドパミン拮抗剤 フェノチアジン系薬剤、ブチロフェノン系薬剤、メトクロプラミド、ドンペリドン | 本剤の作用が減弱するおそれがある。 | 本剤はドパミン作動薬であり、併用により両薬剤の作用が拮抗するおそれがある。 |
| 抗パーキンソン剤 レボドパ、抗コリン剤、アマンタジン塩酸塩、ドロキシドパ | ジスキネジア、幻覚、錯乱等の副作用が増強することがある。 | 相互に作用が増強することがある。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 突発的睡眠(0.1〜5%未満)
前兆のない突発的睡眠があらわれることがある。[1.、8.1参照]
11.1.2 幻覚(15.4%)、妄想(0.1〜5%未満)、せん妄(0.1〜5%未満)、激越(0.1〜5%未満)、錯乱(頻度不明)
11.1.3 抗利尿ホルモン不適合分泌症候群(SIADH)(頻度不明)
低ナトリウム血症、低浸透圧血症、尿中ナトリウム排泄量の増加、高張尿、痙攣、意識障害等を伴う抗利尿ホルモン不適合分泌症候群(SIADH)があらわれることがあるので、異常が認められた場合には投与を中止し、水分摂取の制限等適切な処置を行うこと。
11.1.4 悪性症候群(頻度不明)
パーキンソン病患者において、本剤の急激な減量又は中止により、悪性症候群があらわれることがある。観察を十分に行い、発熱、意識障害、無動無言、高度の筋硬直、不随意運動、嚥下困難、頻脈、血圧の変動、発汗、血清CKの上昇等があらわれた場合には悪性症候群の症状である可能性があるため、再投与後、漸減し、体冷却、水分補給等の適切な処置を行うこと。[8.4参照]
11.1.5 横紋筋融解症(頻度不明)
筋肉痛、脱力感、CK上昇、血中及び尿中ミオグロビン上昇を特徴とする横紋筋融解症があらわれることがある。横紋筋融解症による急性腎障害の発症に注意すること。
11.1.6 肝機能障害(頻度不明)
AST、ALT、LDH、γ-GTP、総ビリルビン上昇等の肝機能障害があらわれることがある。
11.2 その他の副作用
| 5%以上 | 0.1〜5%未満 | 0.1%未満 | 頻度不明 | |
| 過敏症 | 過敏症状 | |||
| 皮膚 | 多汗、蕁麻疹、網状皮斑 | 発疹、そう痒症 | ||
| 筋・骨格系 | CK上昇(7.5%) | 背部痛、腰痛 | ||
| 中枢・末梢神経系 | ジスキネジア(17.5%)、傾眠(16.8%)、めまい(12.5%)、頭痛(5.5%) | ジストニア、緊張亢進、舌麻痺、運動過多、ミオクローヌス、声が出にくい、異常感覚、知覚減退、パーキンソニズムの増悪 | 失神 | |
| 自律神経系 | 口内乾燥(8.3%) | 起立性低血圧、高血圧、唾液増加 | ||
| 感覚器 | 苦味、眼のちらつき、複視、羞明 | 霧視、視力低下 | ||
| 精神神経系 | 食欲不振(12.2%)、不眠(6.5%) | 不安、神経過敏、気分高揚感、悪夢、早朝覚醒、ねぼけ様症状、異夢、徘徊 | 薬剤離脱症候群注)(無感情、不安、うつ、疲労感、発汗、疼痛等)、病的性欲亢進、性欲減退、暴食、病的賭博、不穏、過食(体重増加)、健忘、強迫性購買 | |
| 消化管 | 悪心(29.9%)、消化不良(11.9%)、便秘(9.0%)、胃不快感(6.9%)、嘔吐(5.9%) | 腹痛、胃潰瘍、胃炎、上腹部痛、口内炎、鼓腸放屁、イレウス | 体重減少 | |
| 肝臓 | 肝機能異常(AST上昇、ALT上昇、LDH上昇等) | γ-GTP上昇 | ||
| 内分泌 | プロラクチン低下、成長ホルモン上昇 | |||
| 代謝 | 血糖値上昇 | |||
| 循環器 | 低血圧、動悸 | |||
| 泌尿器系 | 排尿頻回、尿蛋白陽性 | 尿閉 | ||
| 一般的全身障害 | 末梢性浮腫、胸痛、倦怠感、疲労感、脱力感、手がピリピリする、転倒、口渇 | |||
| 呼吸器 | 呼吸困難 | 肺炎、しゃっくり | ||
| 生殖系 | 自発陰茎勃起 |
| 5%未満 | |
| 中枢・末梢神経系 | レストレスレッグス症候群のaugmentation(2.3%) |
13. 過量投与
13.1 症状
悪心、嘔吐、過度の鎮静、運動過多、幻覚、激越、低血圧等の症状を発現する可能性がある。
13.2 処置
精神症状が見られた場合には、抗精神病薬の投与を考慮する。なお、血液透析による除去は期待できない。
14. 適用上の注意
14.1 薬剤交付時の注意
14.1.1 PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
14.1.2 本剤は光に対して不安定なため、服用直前にPTPシートから取り出すよう指導すること。
15. その他の注意
15.1 臨床使用に基づく情報
ヒトにおいて本剤を含む抗パーキンソン剤と網膜変性との関連性は認められなかったとの報告がある。
15.2 非臨床試験に基づく情報
ラットのがん原性試験(24ヶ月間混餌投与)において、2mg/kg/日以上の投与量で網膜変性の増加が報告されている。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
健康成人にプラミペキソール塩酸塩水和物0.1、0.2、0.3mgを空腹時に単回経口投与したときの、血漿中未変化体の薬物動態パラメータを次表に示す。Cmax及びAUCは用量直線性を示した1)。
| 薬物動態パラメータ | 0.1mg | 0.2mg | 0.3mg |
| Cmax(pg/mL) | 294.6±46.3 | 583.2±69.9 | 766.3±88.8 |
| tmax(h) | 1.5±0.5 | 1.4±0.5 | 2.3±1.2 |
| t1/2(h) | 7.71±1.90 | 6.36±1.46 | 6.94±1.09 |
| AUC0-∞(pg・h/mL) | 3139.2±548.5 | 5642.5±681.6 | 9135.8±1422.2 |
16.1.2 反復投与
<血漿中未変化体濃度推移>
健康成人にプラミペキソール塩酸塩水和物0.1mgを第1日目は1日1回、2日目は1日2回、3〜6日目は1日3回、7日目は1回食後反復経口投与したとき、血漿中未変化体濃度は1日3回投与開始後3日で定常状態に達した。また、単回投与後の薬物動態パラメータから予測される以上の反復投与による蓄積性はなかった2)。
<維持量に対する血漿中濃度>
パーキンソン病患者に、プラミペキソール塩酸塩水和物1.0〜4.5mgを反復経口投与後の定常状態(維持量投与開始後4日目以降)において、血漿中濃度のトラフ値は投与量に対して直線的に増加することが示唆された。同一被験者における定常状態のトラフ値に大きな変化は認められず、プラミペキソール塩酸塩水和物反復投与後の定常状態における蓄積は認められなかった3)。
16.1.3 生物学的利用率
16.1.4 生物学的同等性試験
プラミペキソール塩酸塩錠0.5mg「FFP」とビ・シフロール錠0.5mgを、クロスオーバー法によりそれぞれ1錠(プラミペキソール塩酸塩水和物として0.5mg)健康成人男子に絶食時単回経口投与して、血漿中未変化体濃度を測定し、得られた薬物動態パラメータ(AUC、Cmax)について90%信頼区間法にて統計解析を行った結果、log(0.80)〜log(1.25)の範囲内であり、両剤の生物学的同等性が確認された6)。
| 判定パラメータ | 参考パラメータ | |||
| AUC0-24(pg・hr/mL) | Cmax(pg/mL) | Tmax(hr) | t1/2(hr) | |
| プラミペキソール塩酸塩錠0.5mg「FFP」 | 11866±1451 | 1020±178 | 2.8±2.6 | 6.5±0.99 |
| ビ・シフロール錠0.5mg | 12057±1382 | 1011±169 | 2.7±2.3 | 6.6±1.02 |
血漿中濃度並びにAUC、Cmax等のパラメータは、被験者の選択、体液の採取回数・時間等の試験条件によって異なる可能性がある。
16.2 吸収
健康成人にプラミペキソール塩酸塩水和物0.25mgを空腹時又は食後に単回経口投与したときの血漿中未変化体の薬物動態パラメータを比較した。その結果、薬物動態パラメータに有意な差は認められず、プラミペキソール塩酸塩水和物の吸収に対する食事の影響は少ないものと考えられた7)(外国人のデータ)。なお、国内で実施された健康成人に対する単回投与(空腹時投与)及び反復投与の第1日目(食後投与)の薬物動態パラメータを比較した。その結果、tmaxは食後投与で3.1時間と空腹時投与(1.5時間)に比し延長する傾向が認められたが、Cmax、AUC及びt1/2はいずれも類似しており、プラミペキソール塩酸塩水和物の吸収に対する食事の影響は少ないものと考えられた1)2)。
16.3 分布
ヒト血清蛋白結合率は17〜26%であった8)(in vitro)。
16.5 排泄
健康成人に14C-プラミペキソール塩酸塩水和物0.3mgを経口投与したとき、血漿中及び尿中には大部分が未変化体として存在する。また、投与後96時間までに87.6%が尿中に、1.6%が糞中に排泄された。プラミペキソール塩酸塩水和物は尿中排泄が主排泄経路と考えられた4)(外国人のデータ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
健康成人、軽度(50≦クレアチニンクリアランス<80mL/min)、中等度(30≦クレアチニンクリアランス<50mL/min)及び高度(5≦クレアチニンクリアランス<30mL/min)の腎機能障害患者並びに透析患者計26例を対象にプラミペキソール塩酸塩水和物0.25mgを投与し、薬物動態を検討した。その結果、Cmax、tmax及びVd/Fに有意な差は認められなかったが、次表に示すとおりt1/2は中等度及び高度の腎機能障害患者において、健康成人の約3倍に延長した。なお、透析されたプラミペキソール塩酸塩水和物は投与量の約9%であった9)(外国人のデータ)。[7.2、7.3、9.8.2参照]
| 投与対象 | クレアチニンクリアランス(mL/min) | 例数 | AUC0-∞(ng・h/mL) | t1/2(h) | CLtot/F(mL/min) | CLr(mL/min) |
| 健康成人 | >80 | 6 | 7.33±1.49 | 11.3±2.72 | 411±85.9 | 277±59.0 |
| 軽度腎機能障害患者 | 50〜79 | 6 | 10.2±2.29 | 15.3±3.82 | 297±57.2 | 206±79.0注1) |
| 中等度腎機能障害患者 | 30〜49 | 5 | 16.4±5.45 | 36.3±18.8 | 192±52.5 | 105±43.9注2) |
| 高度腎機能障害患者 | 5〜29 | 3 | 22.6±3.48 | 38.4±12.7 | 131±22.2 | 32.8±15.6 |
16.7 薬物相互作用
16.7.1 シメチジン、アマンタジン塩酸塩
健康成人12例を対象にプラミペキソール塩酸塩水和物0.25mg及びシメチジン300mgを併用経口投与し、プラミペキソール塩酸塩水和物の薬物動態に及ぼすシメチジンの影響を検討した。その結果、プラミペキソール塩酸塩水和物単独投与に比し併用投与ではプラミペキソール塩酸塩水和物の尿中未変化体総排泄量に有意な変化は認めなかったが、腎クリアランス(CLr)は30〜39%有意に低下し、t1/2は延長した10)。このことから、プラミペキソール塩酸塩水和物も腎臓の有機カチオン輸送系を介して尿細管分泌されることが示唆された(外国人のデータ)。
また、パーキンソン病患者にプラミペキソール塩酸塩水和物1.0〜4.5mgを反復経口投与し、定常状態(維持量投与開始後4日目以降)における血漿中濃度(52例)から、探索的にポピュレーションファーマコキネティクス解析によりアマンタジン塩酸塩との併用(28例)による影響を検討した結果、プラミペキソール塩酸塩水和物のクリアランスが低下することが確認された11)。[10.2参照]
また、パーキンソン病患者にプラミペキソール塩酸塩水和物1.0〜4.5mgを反復経口投与し、定常状態(維持量投与開始後4日目以降)における血漿中濃度(52例)から、探索的にポピュレーションファーマコキネティクス解析によりアマンタジン塩酸塩との併用(28例)による影響を検討した結果、プラミペキソール塩酸塩水和物のクリアランスが低下することが確認された11)。[10.2参照]
16.8 その他
プラミペキソール塩酸塩錠0.125mg「FFP」は、「含量が異なる経口固形製剤の生物学的同等性試験ガイドライン(平成18年11月24日薬食審査発第1124004号)」に基づき、プラミペキソール塩酸塩錠0.5mg「FFP」を標準製剤としたとき、溶出挙動が等しく、生物学的に同等とみなされた6)。
17. 臨床成績
17.1 有効性及び安全性に関する試験
<パーキンソン病>
17.1.1 国内第III相試験
パーキンソン病患者315例を対象とした国内二重盲検比較試験において、プラミペキソール塩酸塩水和物(0.125mg×2回/日より漸増)、ブロモクリプチンメシル酸塩(1.25mg×1回/日より漸増)又はプラセボを12週間経口投与した時、プラミペキソール塩酸塩水和物はプラセボと比較しUPDRS(Unified Parkinson's Disease Rating Scale)PartII(日常生活動作)及びPartIII(運動能力検査)の各合計スコアを有意に改善した。また、プラミペキソール塩酸塩水和物のスコアの改善はブロモクリプチンメシル酸塩に比較し劣らないことが示された12)13)。
| 投与対象(試験番号) | UPDRS | 投与群a) | 症例数 | 変化量b)(維持期最終時−投与前) | PPX vs PLAC優越性c) | PPX vs BROM非劣性d)変化量差(90%CI) |
| パーキンソン病患者/レボドパ併用(248.505) | PartII | PPX | 102 | −2.50(−3.98) | <0.001 | 0.74(−0.16〜1.63) |
| PLAC | 107 | −1.00 | ||||
| BROM | 104 | (−3.25) | ||||
| PartIII | PPX | 102 | −10.00(−11.75) | <0.001 | 1.76(−0.65〜4.09) | |
| PLAC | 107 | −5.00 | ||||
| BROM | 104 | (−9.98) |
また、UPDRS PartII又はPartIIIの合計スコアが30%以上の減少率を示した症例の割合を次表に示す。
| プラミペキソール塩酸塩水和物 | ブロモクリプチンメシル酸塩 | プラセボ | |
| UPDRS PartII | 56.9%(58/102例) | 49.0%(51/104例) | 29.9%(32/107例) |
| UPDRS PartIII | 63.7%(65/102例) | 60.6%(63/104例) | 36.4%(39/107例) |
プラミペキソール塩酸塩水和物での副作用発現割合は69.6%(71/102例)で、主な副作用は消化不良23.5%(24/102例)、嘔気18.6%(19/102例)、食欲不振16.7%(17/102例)、ジスキネジア15.7%(16/102例)、便秘12.7%(13/102例)、幻覚12.7%(13/102例)、傾眠11.8%(12/102例)、めまい10.8%(11/102例)であった。
17.1.2 海外第III相試験
パーキンソン病患者333例を対象とした海外二重盲検比較試験において、プラミペキソール塩酸塩水和物(0.125mg×3回/日より漸増)又はプラセボを最長32週間経口投与した時、プラミペキソール塩酸塩水和物はプラセボと比較しUPDRS PartII及びPartIIIの各合計スコアを有意に改善した14)15)。
| 投与対象(試験番号) | UPDRS | 投与群a) | 症例数 | 変化量b)(維持期最終時−投与前) | PPX vs PLAC優越性c) | |
| パーキンソン病患者 | レボドパ非併用(248.323) | PartII | PPX | 163 | −1.9 | <0.001 |
| PLAC | 170 | 0.4 | ||||
| PartIII | PPX | 162 | −5.0 | <0.001 | ||
| PLAC | 168 | 0.8 | ||||
プラミペキソール塩酸塩水和物での副作用発現割合は81.1%(133/164例)で、主な副作用は嘔気32.9%(54/164例)、浮動性めまい27.4%(45/164例)、不眠症19.5%(32/164例)、傾眠17.7%(29/164例)、無力症14.0%(23/164例)、便秘12.8%(21/164例)、頭痛12.2%(20/164例)であった。
17.1.3 海外第III相試験
パーキンソン病患者287例を対象とした海外二重盲検比較試験において、プラミペキソール塩酸塩水和物(0.125mg×3回/日より漸増)又はプラセボを最長32週間経口投与した時、プラミペキソール塩酸塩水和物はプラセボと比較しUPDRS PartII及びPartIIIの各合計スコアを有意に改善した16)。
| 投与対象(試験番号) | UPDRS | 投与群a) | 症例数 | 変化量b)(維持期最終時−投与前) | PPX vs PLAC優越性c) | |
| パーキンソン病患者 | レボドパ非併用(248.324) | PartII | PPX | 144 | −2.7 | 0.002 |
| PLAC | 143 | −1.3 | ||||
| PartIII | PPX | 144 | −6.2 | <0.001 | ||
| PLAC | 142 | −2.6 | ||||
プラミペキソール塩酸塩水和物での副作用発現割合は64.0%(94/147例)で、主な副作用は嘔気21.8%(32/147例)、傾眠11.6%(17/147例)、無力症10.9%(16/147例)、浮動性めまい10.2%(15/147例)、起立性低血圧6.8%(10/147例)であった。
17.1.4 海外第III相試験
パーキンソン病患者246例を対象とした海外二重盲検比較試験において、プラミペキソール塩酸塩水和物(0.125mg×3回/日より漸増)、ブロモクリプチンメシル酸塩(1.25mg×1回/日より漸増)又はプラセボを最長9カ月11日間経口投与した時、プラミペキソール塩酸塩水和物はプラセボと比較しUPDRS PartII及びPartIIIの各合計スコアを有意に改善した17)。
| 投与対象(試験番号) | UPDRS | 投与群a) | 症例数 | 変化量b)(維持期最終時−投与前) | PPX vs PLAC優越性c) | |
| パーキンソン病患者 | レボドパ併用(248.326) | PartII | PPX | 79 | −2.50 | <0.001 |
| PLAC | 83 | −0.50 | ||||
| PartIII | PPX | 79 | −6.00 | <0.001 | ||
| PLAC | 83 | −2.00 | ||||
プラミペキソール塩酸塩水和物での副作用発現割合は85.0%(68/80例)で、主な副作用は起立性低血圧35.0%(28/80例)、ジスキネジア33.8%(27/80例)、嘔気27.5%(22/80例)、めまい22.5%(18/80例)、不眠15.0%(12/80例)、錯乱13.8%(11/80例)であった18)。
<中等度から高度の特発性レストレスレッグス症候群(下肢静止不能症候群)>
17.1.5 国内第II相試験
特発性レストレスレッグス症候群患者41例を対象とした国内二重盲検比較試験において、プラミペキソール塩酸塩水和物(0.125mg×1回/日より漸増)又はプラセボを6週間経口投与した時、プラミペキソール塩酸塩水和物はプラセボと比較し終夜睡眠ポリグラフ上で周期性四肢運動指数(PLMI)の有意な減少を示した。また、国際レストレスレッグス症候群研究グループ重症度スケール(IRLS:International Restless Legs Syndrome Study Group Rating Scale)合計スコアのベースラインからの変化量でもプラセボと比較して有意な減少を示した19)。
| 投与群a) | 症例数 | ベースライン | 投与6週 | 変化量b) | PPX vs PLAC優越性c) |
| PPX | 20 | 29.6 | 4.6 | −25.0 | 0.0019 |
| PLAC | 18 | 46.0 | 39.5 | −6.5 |
| 投与群a) | 症例数 | ベースライン | 投与6週 | 変化量b) | PPX vs PLAC優越性c) |
| PPX | 20 | 23.4 | 7.3 | −16.1 | 0.0005 |
| PLAC | 21 | 25.1 | 18.7 | −6.4 |
a)投与群PPX:プラミペキソール塩酸塩水和物、PLAC:プラセボ
b)PLMI、IRLS合計スコア及びベースラインからの変化量は平均値で示した。
c)ANCOVAのp値を示した。
プラミペキソール塩酸塩水和物での副作用発現割合は45.0%(9/20例)で、主な副作用は悪心15.0%(3/20例)、胃不快感15.0%(3/20例)、頭痛10.0%(2/20例)、傾眠10.0%(2/20例)であった。
17.1.6 国内第III相試験
特発性レストレスレッグス症候群患者154例を対象とした国内二重盲検比較試験において、プラミペキソール塩酸塩水和物0.25mg、0.5mg及び0.75mg/日の固定用量を6週間投与し、引き続きプラミペキソール塩酸塩水和物0.25mg〜0.75mgをflexible doseで1日1回46週間投与した時、プラミペキソール塩酸塩水和物0.25mg、0.5mg及び0.75mg/日のいずれの用量群においてもIRLS合計スコアのベースラインからの変化量は10以上減少した。10以上の減少は重症度分類(0-10:軽度、11-20:中等度、21-30:高度、31-40:非常に高度)において重症度が1段階改善することを意味し、臨床的に意義のある改善を示した。また、非盲検期において、投与52週までIRLS合計スコアの安定した減少を示した20)。
| 投与群a) | 症例数 | ベースライン | 投与6週 | 変化量 |
| PPX(合計) | 154 | 22.3 | 10.1 | −12.2 |
| 0.25mg | 48 | 21.4 | 9.8 | −11.7 |
| 0.5mg | 53 | 22.6 | 9.9 | −12.7 |
| 0.75mg | 53 | 22.8 | 10.7 | −12.1 |
| ベースライン | 投与12週 | 投与24週 | 投与52週 | |
| 症例数 | 140 | 138 | 131 | 119 |
| IRLS合計スコア | 22.3 | 8.2 | 7.3 | 4.9 |
| 変化量 | − | −14.1 | −14.9 | −17.2 |
a)投与群PPX:プラミペキソール塩酸塩水和物
IRLS合計スコア及びベースラインからの変化量は平均値で示した。なお、最大0.75mg/日から漸減せずに投与中止した場合でも悪性症候群は認められなかった。
プラミペキソール塩酸塩水和物での副作用発現割合は60.4%(93/154例)で、主な副作用は悪心31.8%(49/154例)、傾眠17.5%(27/154例)、頭痛7.1%(11/154例)、胃不快感5.8%(9/154例)、便秘5.2%(8/154例)、嘔吐5.2%(8/154例)であった。
18. 薬効薬理
18.1 作用機序
18.1.1 ドパミンD2受容体に対する親和性(in vitro)
18.1.2 ドパミンD2受容体刺激作用
18.2 パーキンソン病様症状改善作用
18.2.1 MPTP誘発症状改善作用
MPTP誘発パーキンソン病様症状をブロモクリプチンメシル酸塩より低用量で改善した(アカゲザル)25)。
18.2.2 無動・固縮に対する改善作用
レセルピン誘発無動・固縮症状の改善作用を示した。これらの改善作用はレボドパとの併用により増強することが認められた(マウス)25)。
18.3 レストレスレッグス症候群様症状の改善作用
6-OHDA(6-hydroxydopamine)により脳内のドパミン神経を変性させたラットにおいては、立ち上がり行動回数と立位時間が増加したが、プラミペキソール塩酸塩水和物投与によりこれらの増加は抑制された26)。
19. 有効成分に関する理化学的知見
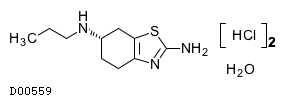
19.1. プラミペキソール塩酸塩水和物
| 一般的名称 | プラミペキソール塩酸塩水和物 |
|---|---|
| 一般的名称(欧名) | Pramipexole Hydrochloride Hydrate |
| 化学名 | (S)-2-Amino-4,5,6,7-tetrahydro-6-propylaminobenzothiazole dihydrochloride monohydrate |
| 分子式 | C10H17N3S・2HCl・H2O |
| 分子量 | 302.26 |
| 融点 | 約290℃(分解) |
| 物理化学的性状 | 白色〜微黄白色の結晶又は結晶性の粉末である。 水に極めて溶けやすく、メタノールにやや溶けやすく、エタノール(95)にやや溶けにくく、テトラヒドロフランにほとんど溶けない。 |
| KEGG DRUG |
|
20. 取扱い上の注意
アルミピロー包装開封後は、湿気を避けて遮光して保存すること。
22. 包装
<プラミペキソール塩酸塩錠0.125mg「FFP」>
(PTP)100錠(10錠×10)
<プラミペキソール塩酸塩錠0.5mg「FFP」>
(PTP)100錠(10錠×10)
23. 主要文献
- 入江伸 他, 臨床医薬, 19 (2), 149-161, (2003)
- 入江伸 他, 臨床医薬, 19 (2), 163-177, (2003)
- 日本人パーキンソン病患者の薬物動態解析(ビ・シフロール錠:2003年12月2日承認、申請資料概要ヘ.3.(2))
- 外国健康成人の薬物動態解析(標識)(ビ・シフロール錠:2003年12月2日承認、申請資料概要ヘ.3.(1))
- 外国健康成人の薬物動態解析(非標識)(ビ・シフロール錠:2003年12月2日承認、申請資料概要ヘ.3.(1))
- 共創未来ファーマ株式会社 社内資料:生物学的同等性試験
- 薬物動態に対する食事の影響(ビ・シフロール錠:2003年12月2日承認、申請資料概要ヘ.3.(1))
- Yokoyama,K.et al., 薬物動態, 14 (4), 300-308, (1999) »DOI
- 外国人腎機能障害患者の薬物動態解析(ビ・シフロール錠:2003年12月2日承認、申請資料概要ヘ.3.(2))
- 薬物動態に対するシメチジンの影響(ビ・シフロール錠:2003年12月2日承認、申請資料概要ヘ.3.(3))
- 日本人パーキンソン病患者母集団動態解析(ビ・シフロール錠:2003年12月2日承認、申請資料概要ヘ.3.(2))
- Mizuno,Y.et al., Movement Disorders., 18 (10), 1149-1156, (2003) »PubMed
- 日本での第III相比較試験/L-DOPA併用(試験248.505)(ビ・シフロール錠:2003年12月2日承認、申請資料概要ト.3.(3).1))
- Shannon,K.M., Neurology., 49 (3), 724-728, (1997) »PubMed
- 欧米での第III相比較試験/L-DOPA非併用(試験248.323)(ビ・シフロール錠:2003年12月2日承認、申請資料概要ト.3.(4).1))
- 外国人初期パーキンソン病患者第III相試験(ビ・シフロール錠:2003年12月2日承認、申請資料概要ト.3.(4).2))
- Guttman,M., Neurology., 49 (4), 1060-1065, (1997) »PubMed
- 欧州での第III相比較試験L-DOPA併用(試験248.326)(ビ・シフロール錠:2003年12月2日承認、申請資料概要ト.3.(4).2))
- 日本人特発性レストレスレッグス症候群患者第II相試験(ビ・シフロール錠:2010年1月20日承認、申請資料概要2.7.6.1)
- 日本人特発性レストレスレッグス症候群患者第III相試験(ビ・シフロール錠:2010年1月20日承認、申請資料概要2.7.6.2)
- Mierau,J.et al., Eur.J.Pharmacol., 290, 29-36, (1995) »PubMed
- Mierau,J., Clin.Neuropharmacol., 18, S195-S206, (1995)
- Domino,E.F.et al., Eur.J.Pharmacol., 325, 137-144, (1997) »PubMed
- 作用機序に関する薬理学的検討(ビ・シフロール錠:2003年12月2日承認、申請資料概要ホ-1)
- 武内正吾 他, 医学と薬学, 49 (6), 973-983, (2003)
- Ondo,W.G.et al., Movement Disorders., 15 (1), 154-158, (2000) »PubMed
24. 文献請求先及び問い合わせ先
文献請求先
共創未来ファーマ株式会社
お客様相談室
〒155-8655
東京都世田谷区代沢5-2-1
電話:050-3383-3846
製品情報問い合わせ先
共創未来ファーマ株式会社
お客様相談室
〒155-8655
東京都世田谷区代沢5-2-1
電話:050-3383-3846
26. 製造販売業者等
26.1 製造販売元
共創未来ファーマ株式会社
東京都品川区広町1-4-4