医薬品情報
| 総称名 | テイコプラニン |
|---|---|
| 一般名 | テイコプラニン |
| 欧文一般名 | Teicoplanin |
| 製剤名 | 注射用テイコプラニン |
| 薬効分類名 | グリコペプチド系抗生物質製剤 |
| 薬効分類番号 | 6119 |
| ATCコード | J01XA02 |
| KEGG DRUG |
D02142
テイコプラニン
|
| KEGG DGROUP |
DG01580
グリコペプチド系抗生物質
|
| JAPIC | 添付文書(PDF) |
添付文書情報2023年6月 改訂(第1版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| テイコプラニン点滴静注用200mg「明治」 (後発品) | TEICOPLANIN for I.V.Infusion「MEIJI」 | Meiji Seikaファルマ | 6119401D1159 | 1337円/瓶 | 劇薬, 処方箋医薬品注) |
| テイコプラニン点滴静注用400mg「明治」 (後発品) | TEICOPLANIN for I.V.Infusion「MEIJI」 | Meiji Seikaファルマ | 6119401D2058 | 2492円/瓶 | 劇薬, 処方箋医薬品注) |
2. 禁忌
次の患者には投与しないこと
本剤の成分に対し過敏症の既往歴のある患者
4. 効能または効果
5. 効能または効果に関連する注意
本剤はメチシリン耐性の黄色ブドウ球菌感染症に対してのみ有用性が認められている。
6. 用法及び用量
通常、成人にはテイコプラニンとして初日400mg(力価)又は800mg(力価)を2回に分け、以後1日1回200mg(力価)又は400mg(力価)を30分以上かけて点滴静注する。敗血症には、初日800mg(力価)を2回に分け、以後1日1回400mg(力価)を30分以上かけて点滴静注する。
通常、乳児、幼児又は小児にはテイコプラニンとして10mg(力価)/kgを12時間間隔で3回、以後6〜10mg(力価)/kg(敗血症などの重症感染症では10mg(力価)/kg)を24時間ごとに30分以上かけて点滴静注する。また、新生児(低出生体重児を含む)にはテイコプラニンとして初回のみ16mg(力価)/kgを、以後8mg(力価)/kgを24時間ごとに30分以上かけて点滴静注する。
なお、年齢、体重、症状により適宜増減する。
7. 用法及び用量に関連する注意
7.1 投与期間中は血中濃度をモニタリングすることが望ましい。トラフレベルの血中濃度は5〜10μg/mLを保つことが投与の目安となるが、敗血症などの重症感染症においては確実な臨床効果を得るために10μg/mL以上を保つこと。[9.1.2、9.2.1、9.2.2、9.7、9.8参照]
7.2 本剤は主として腎臓から排泄され、腎機能障害患者では腎機能正常者よりも血中半減期が延長するので、投与量を調節して使用する必要がある。クレアチニン・クリアランスから投与量又は投与間隔を調節する目安は以下のとおりである。なお、血液透析あるいは腹膜透析を受けている患者への投与は、クレアチニン・クリアランスが10mL/min以下の患者と同様とする。[9.2.1、9.2.2、16.6.1参照]
| 障害度 | 初期投与(3日目まで) | 4日目以降 |
| 60≧Ccr>40 | 腎機能正常者と等しい投与量 | 1日の用量を半減するかあるいは隔日に投与する。 |
| 40≧Ccr>10 | 腎機能正常者と等しい投与量 | 1日の用量を1/3に減ずるかあるいは3日ごとに投与する。 |
| 10≧Ccr | 腎機能正常者と等しい投与量 | 1日の用量を1/5に減ずるかあるいは5日ごとに投与する。 |
7.3 ショック及びレッドマン症候群(顔、頸、躯幹の紅斑性充血、そう痒等)が報告されているので、本剤の使用にあたっては30分以上かけて点滴静注し、急速なワンショット静注では使用しないこと。[11.1.1参照]
8. 重要な基本的注意
8.1 本剤によるショック、アナフィラキシーの発生を確実に予知できる方法がないので、次の措置をとること。[11.1.1参照]
8.1.1 事前に既往歴等について十分な問診を行うこと。なお、抗生物質等によるアレルギー歴は必ず確認すること。
8.1.2 投与に際しては、必ずショック等に対する救急処置のとれる準備をしておくこと。
8.1.3 投与開始から投与終了後まで、患者を安静の状態に保たせ、十分な観察を行うこと。特に、投与開始直後は注意深く観察すること。
8.2 本剤の使用にあたっては、耐性菌の発現を防ぐため、原則として感受性を確認し、疾病の治療上必要な最小限の期間の投与にとどめること。
8.3 眩暈、耳鳴、聴力低下等の第8脳神経障害があらわれることがあるので、聴力検査を行う等観察を十分に行うこと。[11.1.2参照]
8.4 無顆粒球症、白血球減少、血小板減少があらわれることがあるので、定期的に検査を行うなど観察を十分に行うこと。[11.1.4参照]
8.5 急性腎障害があらわれることがあるので、定期的に検査を行うなど観察を十分に行うこと。[11.1.5参照]
8.6 AST、ALT、LDH、Al-P、γ-GTP、総ビリルビン等の上昇、黄疸があらわれることがあるので、定期的に検査を行うなど観察を十分に行うこと。[11.1.6参照]
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 アミノグリコシド系抗生物質、ペプチド系抗生物質又はバンコマイシン類に対し過敏症の既往歴のある患者(ただし、テイコプラニンに対し過敏症の既往歴のある患者には投与しないこと)
9.1.2 アミノグリコシド系抗生物質、グリコペプチド系抗生物質又はバンコマイシン類による難聴又はその他の難聴のある患者
9.2 腎機能障害患者
9.2.1 血液透析患者
9.2.2 腎機能障害のある患者(血液透析患者を除く)
9.3 肝機能障害患者
肝障害を悪化させることがある。[11.1.6参照]
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上まわると判断される場合にのみ投与すること。
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。動物実験(ラット)で乳汁中に移行することが認められている。
9.7 小児等
9.8 高齢者
血中濃度をモニタリングするなど安全性の確保に配慮すること。また、投与前及び投与中に腎機能検査を行い、腎機能の低下の程度により、4日目以降の用量を減量するなど慎重に投与すること。腎機能が低下している場合が多い。[7.1参照]
10. 相互作用
10.2 併用注意
| ループ利尿剤 エタクリン酸 フロセミド 等 [9.2.1、9.2.2参照] | 腎障害、聴覚障害を増強するおそれがあるので併用は避けることが望ましいが、やむを得ず併用する場合は、血中濃度をモニタリングするなど安全性の確保に配慮し、慎重に投与すること。 | 腎障害、聴覚毒性が増強される。 |
| 腎障害、聴覚障害を起こす可能性のある薬剤 アミノグリコシド系抗生物質 アルベカシン ゲンタマイシン イセパマイシン 等 ペプチド系抗生物質 バンコマイシン アムホテリシンB シクロスポリン シスプラチン 等 [9.1.2、9.2.1、9.2.2参照] | 腎障害、聴覚障害を増強するおそれがあるので併用は避けることが望ましいが、やむを得ず併用する場合は、血中濃度をモニタリングするなど安全性の確保に配慮し、慎重に投与すること。 | 腎障害、聴覚毒性が増強される。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 ショック、アナフィラキシー(いずれも頻度不明)
11.1.2 第8脳神経障害(頻度不明)
眩暈、耳鳴、聴力低下等の第8脳神経障害があらわれることがある。[8.3参照]
11.1.3 中毒性表皮壊死融解症(Toxic Epidermal Necrolysis:TEN)、皮膚粘膜眼症候群(Stevens-Johnson症候群)、急性汎発性発疹性膿疱症、紅皮症(剥脱性皮膚炎)(いずれも頻度不明)
11.1.4 無顆粒球症、白血球減少、血小板減少(いずれも頻度不明)[8.4参照]
11.1.5 急性腎障害(頻度不明)[8.5参照]
11.1.6 肝機能障害、黄疸(いずれも頻度不明)
11.2 その他の副作用
| 1〜5%未満注1) | 0.1〜1%未満 | 0.1%未満 | 頻度不明 | |
| 過敏症注2) | 発熱、発疹 | |||
| 肝臓 | AST上昇、ALT上昇、Al-P上昇、γ-GTP上昇 | 黄疸、LDH上昇、ビリルビン上昇 | ||
| 血液 | 好酸球増多 | 貧血、白血球減少 | 汎血球減少 | |
| 腎臓 | BUN上昇 | 血清クレアチニン上昇 | ||
| 循環器注3) | 血圧低下 | 動悸 | 血圧上昇 | |
| 消化器 | 食欲不振、下痢、嘔吐 | 悪心 | ||
| その他 | 痙攣 | 注射部位疼痛、静脈炎、悪寒、頭痛、菌交代症 |
14. 適用上の注意
14.1 薬剤調製時の注意
14.1.1 注射液の調製にあたっては、200mg製剤1バイアル[200mg(力価)]には注射用水又は生理食塩液約5mL、400mg製剤1バイアル[400mg(力価)]には注射用水又は生理食塩液約10mLを加えてなるべく泡立たないように穏やかに溶解し溶液とする。この溶解液を100mL以上の生理食塩液等に加えて希釈する。なお、新生児、乳児、幼児及び小児においては、注射用水又は生理食塩液を、200mg製剤1バイアルには5mL、400mg製剤1バイアルには10mL加えた溶解液から投与量相当分を採取し、生理食塩液等にて適宜希釈して調製する。
14.1.2 大塚糖液5%、マルトス輸液10%との配合については、調製後、速やかに使用すること。
14.1.3 調製後は速やかに使用し、残液は廃棄すること。
14.2 薬剤投与時の注意
14.2.1 乾燥ポリエチレングリコール処理人免疫グロブリン、ポリエチレングリコール処理人免疫グロブリン、ガベキサートメシル酸塩、アムホテリシンB、ミノサイクリン塩酸塩と配合すると白濁・沈殿を生じることが確認されているので、これらの薬剤とは混注しないこと。
14.2.2 セフォチアムと混合すると、本剤の活性低下を来すことが確認されているので、併用する場合には別々に投与すること。
15. その他の注意
15.1 臨床使用に基づく情報
15.1.1 米国において感染性心内膜炎・敗血症及び骨・関節感染症を対象とし、高用量を用いた臨床試験〔投与量:6〜30mg/kg(400〜2,000mg)を初日は2回、2日目以降1日1回〕で、トラフレベルの血中濃度が60μg/mL以上を示した症例に血清クレアチニンの異常変動の発現頻度が高かったことから、トラフレベルの血中濃度が60μg/mL以上になった場合には腎障害・聴覚障害等の副作用の発現に注意すること。
また、トラフレベルの血中濃度が20μg/mL以上で、一過性に肝機能検査値が軽度上昇したとの報告がある。
また、トラフレベルの血中濃度が20μg/mL以上で、一過性に肝機能検査値が軽度上昇したとの報告がある。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与(成人)
健康成人男子各群5例に2、4及び8mg/kgのテイコプラニンを30分かけて点滴静注したときの最高血漿中濃度はそれぞれ17.0、34.4及び71.8μg/mLを示し、投与後初期に比較的速やかに減少した後、終末半減期46〜56時間ときわめて穏やかに消失した1)。
図 健康成人における用量別血漿中テイコプラニン濃度
(平均値±標準偏差、n=5)
16.1.2 反復投与(成人)
健康成人男子5例にテイコプラニンを、4mg/kg1日1回7日間反復静脈内投与(試験A)、及び、健康成人男子3例に初日400mg2回、2〜5日目は1日1回400mgを反復静脈内投与(試験B)し、血漿中濃度を調べた1)。
試験Aにおいては、投与後24時間値(トラフ濃度)は2.5μg/mLであった。反復投与5〜6回でほぼ定常状態に達し、最終投与後24時間値は初回投与時の約3倍(7.2μg/mL)となった。
試験Bにおいては、初回投与12時間後のloading doseによりトラフ濃度は速やかに定常状態に達し、以後反復投与により10〜12μg/mLのトラフ濃度が維持された。
試験Aにおいては、投与後24時間値(トラフ濃度)は2.5μg/mLであった。反復投与5〜6回でほぼ定常状態に達し、最終投与後24時間値は初回投与時の約3倍(7.2μg/mL)となった。
試験Bにおいては、初回投与12時間後のloading doseによりトラフ濃度は速やかに定常状態に達し、以後反復投与により10〜12μg/mLのトラフ濃度が維持された。
16.1.3 反復投与(小児)
16.3 分布
16.3.1 ヒトにテイコプラニンを静注又は点滴静注したとき、心臓組織3)、皮下脂肪4)、水疱液5)、骨組織6)、滑液6)、肺組織6)及び気管支分泌物6)への移行は良好で1〜2μg/mL(又はg)以上であった(外国人データ)。
16.3.2 血清蛋白結合率
ヒト血清蛋白質への結合率は約90%である7)(外国人のデータ)。
16.3.3 アルブミンとの結合
ヒト血清アルブミン−ビリルビン結合に対するテイコプラニンのビリルビン遊離作用を検討したとき、テイコプラニンのビリルビン遊離作用は認められなかった8)(in vitro)。
16.5 排泄
健康成人男子各群5例に2、4及び8mg/kgのテイコプラニンを30分かけて点滴静注したとき、投与開始後96時間までの尿中排泄率は投与量の46〜54%であった。8mg/kg投与後3日間の糞中排泄は平均1%未満であった1)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
外国人腎機能障害患者に3mg/kgのテイコプラニンを投与したとき、投与後初期の血漿中濃度に健康成人と差は見られないが、クレアチニン・クリアランスの低下に相関して消失半減期が延長するとの報告がある。したがって腎機能障害患者においては投与間隔あるいは投与量の調節が必要である10)。[7.2、9.2.1、9.2.2参照]
| グループ | |||||
| I | II | III | IV | V | |
| (健康成人) | (腎機能障害患者) | (CAPD患者) | |||
| クレアチニン・クリアランス Ccr(mL/min) | 103.0±2.4 | 45.7±11.5 | 16.8±3.2 | 6.9±2.3 | ≦2 |
| 分布容積 Vdss(L/kg) | 0.84±0.17 | 0.94±0.22 | 0.99±0.18 | 1.01±0.33 | 1.00±0.22 |
| 全身クリアランス CLt(mL/min) | 18.1±3.4 | 10.3±2.1 | 10.2±2.3 | 6.3±1.6 | 5.6±2.0 |
注)93日齢の低出生体重児を含む。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国内臨床試験
本剤の臨床試験はオープン試験で実施され、投与量は1日維持用量200mg(力価)又は400mg(力価)を重症度で選択した。MRSA感染症(成人)における有効性評価対象症例は43例であり、有効率は79.1%であった。
| 疾患名 | 有効率(%) |
| 敗血症 | 6/10(60.0) |
| 深在性皮膚感染症(せつ・せつ腫症・よう) | 15/16(93.8) |
| 慢性膿皮症(皮下膿瘍・膿皮症) | |
| 外傷・熱傷及び手術創等の二次感染 | |
| 肺炎 | 13/17(76.5) |
| 膿胸 | |
| 慢性呼吸器病変の二次感染(慢性気管支炎) | |
| (著効+有効)/症例数 | 34/43(79.1) |
17.2 製造販売後調査等
17.2.1 小児
(1)小児及び新生児を対象とした市販後臨床試験において、維持用量を小児10mg/kg/日、新生児8mg/kg/日として投与を行った。小児(41日齢〜10歳)では8例中5例、新生児注)(4日齢〜93日齢)では9例中8例、合計76.5%(13/17例)の有効率(全般的改善度)であった。
本剤と因果関係の否定できない有害事象は17例中3例(17.6%)、5件にみとめられた2)。
本剤と因果関係の否定できない有害事象は17例中3例(17.6%)、5件にみとめられた2)。
(2)小児及び新生児を対象とした特別調査を行った結果、各種MRSA感染症と推定される小児及び新生児(0日齢〜15歳)に対して、有効率(全般的改善度)は88.5%(23/26例)であった。また、菌消失率は75.0%(18/24例)であった。
副作用は9例(20.0%)で、主な副作用は肝機能検査値の異常であった16)。
副作用は9例(20.0%)で、主な副作用は肝機能検査値の異常であった16)。
注)93日齢の低出生体重児を含む。
18. 薬効薬理
18.1 作用機序
本剤の作用は、細菌の細胞壁合成阻害によるものであり、その作用は殺菌的である17)。
18.2 抗菌作用
18.3 耐性
MRSAを用いたin vitroでの耐性獲得の継代培養試験により検討した結果、耐性化は低い22)。
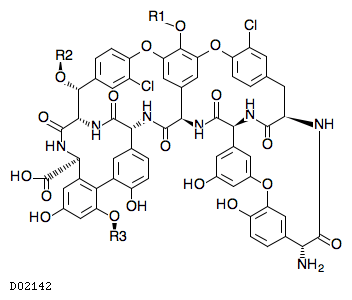
19. 有効成分に関する理化学的知見
19.1. テイコプラニン
| 一般的名称 | テイコプラニン |
|---|---|
| 一般的名称(欧名) | Teicoplanin |
| 化学名 | テイコプラニンA2-1、テイコプラニンA2-2、テイコプラニンA2-3、テイコプラニンA2-4、テイコプラニンA2-5及びテイコプラニンA3-1等の混合物 |
| 分子式 | C72〜89H68〜99Cl2N8〜9O28〜33 |
| 分子量 | 1564.25〜1893.68 |
| 物理化学的性状 | テイコプラニンは白色〜淡黄白色の粉末である。 本品は水に溶けやすく、N,N-ジメチルホルムアミドにやや溶けにくく、アセトニトリル、メタノール、エタノール(95)、アセトン、酢酸(100)又はジエチルエーテルにほとんど溶けない。 |
| KEGG DRUG |
|
21. 承認条件
本剤使用後の本剤耐性及びバンコマイシン耐性菌の出現状態を十分に調査し、医療関係者に情報提供すること。
22. 包装
<テイコプラニン点滴静注用200mg「明治」>
10バイアル
<テイコプラニン点滴静注用400mg「明治」>
10バイアル
23. 主要文献
- 中島光好 他, 日本化学療法学会雑誌, 41 (S-2), 88-102, (1993)
- 砂川慶介 他, Jpn.J.Antibiot., 55 (5), 656-677, (2002) »PubMed
- Bergeron,M.G.,et al., Antimicrob.Agents Chemother., 34 (9), 1699-1702, (1990) »PubMed
- Antrum,R.M.,et al., Drugs Exp.Clin.Res., 15 (1), 21-23, (1989) »PubMed
- Novelli,A.,et al., Int.J.Clin.Pharm.Res., 9 (3), 233-237, (1989) »PubMed
- 第十八改正日本薬局方解説書, C-3285-C-3294, (2021)
- Assandri,A.,et al., Eur.J.Clin.Pharmacol., 33 (2), 191-195, (1987) »PubMed
- 注射用タゴシッド:2003年1月31日承認、申請資料概要ヘ.2.(3)
- Bernareggi,A.,et al., Antimicrob.Agents Chemother., 30 (5), 733-738, (1986) »PubMed
- Bonati,M.,et al., Clin.Pharmacokinet., 12 (4), 292-301, (1987) »PubMed
- 副島林造, 日本化学療法学会雑誌, 41 (S-2), 115-125, (1993)
- 伊藤 章 他, 日本化学療法学会雑誌, 41 (S-2), 126-133, (1993)
- 由良二郎 他, 日本化学療法学会雑誌, 41 (S-2), 134-145, (1993)
- 注射用タゴシッド:2003年1月31日承認、申請資料概要イ.2.(1)
- 注射用タゴシッド:2003年1月31日承認、申請資料概要 効能・効果の設定理由
- 注射用タゴシッド:2003年1月31日承認、申請資料概要ト.1.(2)
- Somma,S.,et al., Antimicrob.Agents Chemother., 26 (6), 917-923, (1984) »PubMed
- 井上松久 他, 日本化学療法学会雑誌, 41 (S-2), 47-55, (1993)
- 出口浩一 他, 日本化学療法学会雑誌, 41 (S-2), 32-40, (1993)
- 五島瑳智子 他, 日本化学療法学会雑誌, 41 (S-2), 18-24, (1993)
- 加藤直樹 他, 日本化学療法学会雑誌, 41 (S-2), 56-61, (1993)
- 中塩哲士 他, 日本化学療法学会雑誌, 41 (S-2), 41-46, (1993)
24. 文献請求先及び問い合わせ先
文献請求先
Meiji Seikaファルマ株式会社
くすり相談室
〒104-8002
東京都中央区京橋2-4-16
電話:フリーダイヤル0120-093-396
03-3273-3539
03-3273-3539
FAX:03-3272-2438
製品情報問い合わせ先
Meiji Seikaファルマ株式会社
くすり相談室
〒104-8002
東京都中央区京橋2-4-16
電話:フリーダイヤル0120-093-396
03-3273-3539
03-3273-3539
FAX:03-3272-2438
26. 製造販売業者等
26.1 製造販売元
Meiji Seikaファルマ株式会社
東京都中央区京橋2-4-16