医薬品情報
| 総称名 | ジャカビ |
|---|---|
| 一般名 | ルキソリチニブリン酸塩 |
| 欧文一般名 | Ruxolitinib Phosphate |
| 製剤名 | ルキソリチニブリン酸塩製剤 |
| 薬効分類名 | ヤヌスキナーゼ(JAK)阻害剤 |
| 薬効分類番号 | 3999 4291 |
| ATCコード | L01EJ01 |
| KEGG DRUG |
D09960
ルキソリチニブリン酸塩
|
| KEGG DGROUP |
DG02020
JAK阻害薬
DG01985
疾患修飾性抗リウマチ薬 (DMARD)
|
| JAPIC | 添付文書(PDF) |
添付文書情報2025年2月 改訂(第5版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ジャカビ錠5mg | JAKAVI Tablets | ノバルティスファーマ | 4291034F1029 | 3636.6円/錠 | 劇薬, 処方箋医薬品 |
| ジャカビ錠10mg | JAKAVI Tablets | ノバルティスファーマ | 4291034F2025 | 7306.3円/錠 | 劇薬, 処方箋医薬品 |
| ジャカビ内用液小児用0.5% | JAKAVI Oral Solution | ノバルティスファーマ | 4291034S1027 | 12209円/mL | 劇薬, 処方箋医薬品 |
1. 警告
2. 禁忌
次の患者には投与しないこと
2.1 本剤の成分に対し過敏症の既往歴のある患者
2.2 妊婦又は妊娠している可能性のある女性[9.5参照]
4. 効能または効果
5. 効能または効果に関連する注意
ジャカビ錠5mg
<骨髄線維症>
5.1 患者のリスク分類、脾臓の大きさ等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分理解した上で、適応患者の選択を行うこと。
5.2 病理組織学的検査を行い、骨髄線維症と診断された患者に使用すること。
<真性多血症>
5.3 ヒドロキシカルバミドによる適切な治療を行っても十分な効果が認められない場合、又はヒドロキシカルバミドによる治療が不適当と判断される場合に本剤の投与を考慮すること。
5.4 臨床試験に組み入れられた患者の脾臓の大きさ等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分理解した上で、適応患者の選択を行うこと。
<造血幹細胞移植後の移植片対宿主病>
5.5 臨床試験に組み入れられた患者の移植片対宿主病の重症度等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、適応患者の選択を行うこと。
ジャカビ錠10mg
<骨髄線維症>
5.1 患者のリスク分類、脾臓の大きさ等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分理解した上で、適応患者の選択を行うこと。
5.2 病理組織学的検査を行い、骨髄線維症と診断された患者に使用すること。
<真性多血症>
5.3 ヒドロキシカルバミドによる適切な治療を行っても十分な効果が認められない場合、又はヒドロキシカルバミドによる治療が不適当と判断される場合に本剤の投与を考慮すること。
5.4 臨床試験に組み入れられた患者の脾臓の大きさ等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分理解した上で、適応患者の選択を行うこと。
<造血幹細胞移植後の移植片対宿主病>
5.5 臨床試験に組み入れられた患者の移植片対宿主病の重症度等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、適応患者の選択を行うこと。
ジャカビ内用液小児用0.5%
<造血幹細胞移植後の移植片対宿主病>
5.5 臨床試験に組み入れられた患者の移植片対宿主病の重症度等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、適応患者の選択を行うこと。
6. 用法及び用量
ジャカビ錠5mg
錠5mg
<骨髄線維症>
通常、成人には本剤を1日2回、12時間毎を目安に経口投与する。用量は、ルキソリチニブとして1回5mg〜25mgの範囲とし、患者の状態により適宜増減する。
<真性多血症>
通常、成人にはルキソリチニブとして1回10mgを開始用量とし、1日2回、12時間毎を目安に経口投与する。患者の状態により適宜増減するが、1回25mg1日2回を超えないこと。
<造血幹細胞移植後の移植片対宿主病>
通常、成人及び12歳以上の小児にはルキソリチニブとして1回10mgを1日2回、12時間毎を目安に経口投与する。患者の状態により適宜減量する。
通常、6歳以上12歳未満の小児にはルキソリチニブとして1回5mgを1日2回、12時間毎を目安に経口投与する。患者の状態により適宜減量する。
ジャカビ錠10mg
錠10mg
<骨髄線維症>
通常、成人には本剤を1日2回、12時間毎を目安に経口投与する。用量は、ルキソリチニブとして1回5mg〜25mgの範囲とし、患者の状態により適宜増減する。
<真性多血症>
通常、成人にはルキソリチニブとして1回10mgを開始用量とし、1日2回、12時間毎を目安に経口投与する。患者の状態により適宜増減するが、1回25mg1日2回を超えないこと。
<造血幹細胞移植後の移植片対宿主病>
通常、成人及び12歳以上の小児にはルキソリチニブとして1回10mgを1日2回、12時間毎を目安に経口投与する。患者の状態により適宜減量する。
通常、6歳以上12歳未満の小児にはルキソリチニブとして1回5mgを1日2回、12時間毎を目安に経口投与する。患者の状態により適宜減量する。
ジャカビ内用液小児用0.5%
内用液
<造血幹細胞移植後の移植片対宿主病>
通常、6歳以上12歳未満の小児にはルキソリチニブとして1回5mgを1日2回、12時間毎を目安に経口投与する。患者の状態により適宜減量する。
通常、6歳未満の小児にはルキソリチニブとして1回4mg/m2を1日2回、12時間毎を目安に経口投与する。患者の状態により適宜減量する。
7. 用法及び用量に関連する注意
ジャカビ錠5mg
<骨髄線維症、真性多血症>
7.1 他の抗悪性腫瘍剤との併用について、有効性及び安全性は確立していない。
7.2 十分な効果が認められず、血球数から増量可能と判断できる場合は、1回の投与量を5mgずつ2週間以上の間隔をあけて増量することができる。ただし、本剤の初回投与後、4週間は増量しないこと。
<骨髄線維症>
7.3 本剤の投与開始にあたっては、血小板数に基づき次表を参考に開始用量を決定すること。
| 血小板数注) | 開始用量 |
| 20万/mm3超 | 1回20mg1日2回 |
| 10万/mm3以上20万/mm3以下 | 1回15mg1日2回 |
7.4 本剤の投与中に血小板数が減少した場合、下表を参考に減量又は休薬を考慮すること。なお、血小板数が休薬前の数値以上に回復した場合には、1回5mg1日2回から投与を再開できる。ただし、患者の状態をより慎重に観察し、有害事象の発現に十分注意すること。
| 血小板数 | 1回あたりの用量(1日2回) | ||||
| 25mg | 20mg | 15mg | 10mg | 5mg | |
| 10万/mm3以上12.5万/mm3未満 | 20mg | 変更なし | |||
| 7.5万/mm3以上10万/mm3未満 | 10mg | 10mg | 10mg | 変更なし | |
| 5万/mm3以上7.5万/mm3未満 | 5mg | 5mg | 5mg | 5mg | 変更なし |
| 5万/mm3未満 | 休薬 | ||||
7.5 本剤の投与中に好中球数が500/mm3未満に減少した場合には休薬すること。なお、好中球数が休薬前の数値以上に回復した場合には、1回5mg1日2回から投与を再開できる。ただし、患者の状態をより慎重に観察し、有害事象の発現に十分注意すること。
<真性多血症>
7.6 血小板数が5万/mm3以上10万/mm3未満の患者における開始用量の情報は得られていないため、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分理解した上で、本剤の投与の可否を慎重に検討すること。血小板数5万/mm3以上10万/mm3未満の患者に投与可能と判断する場合、低用量から投与を開始するとともに、観察を十分に行い、有害事象の発現に十分注意すること。血小板数5万/mm3未満の患者に対する投与は避けること。
7.7 本剤の投与中に血小板数又はヘモグロビンが減少した場合、下表を参考に減量又は休薬を考慮すること。減量幅は、1回の投与量として5mgとする。なお、血小板数及びヘモグロビンが休薬前の数値以上に回復した場合には、1回5mg1日2回から投与を再開できる。ただし、患者の状態をより慎重に観察し、有害事象の発現に十分注意すること。
| 血小板数 | 5万/mm3以上、10万/mm3未満 | 減量 |
| 5万/mm3未満 | 休薬 | |
| ヘモグロビン | 8g/dL以上、12g/dL未満 | 減量 |
| 8g/dL未満 | 休薬 |
7.8 本剤の投与中に好中球数が1,000/mm3未満に減少した場合には休薬すること。なお、好中球数が休薬前の数値以上に回復した場合には、1回5mg1日2回から投与を再開できる。ただし、患者の状態をより慎重に観察し、有害事象の発現に十分注意すること。
<造血幹細胞移植後の移植片対宿主病>
7.9 副作用により本剤を休薬、減量する場合は、以下の基準を考慮すること。[9.1.4参照]
| 投与量 | 減量又は休薬 |
| 1回10mg1日2回 | 1回5mg1日2回 |
| 1回5mg1日2回 | 1回5mg1日1回 |
| 1回5mg1日1回 | 休薬 |
| 1回4mg/m21日2回 | 1回2mg/m21日2回 |
| 1回2mg/m21日2回 | 休薬 |
| 血小板数 | |
| 1.5万/mm3以上2万/mm3未満 | 1段階減量する。減量後7日以内に2万/mm3以上に回復した場合は、減量前の用量を再開してもよい。 減量後7日を過ぎても2万/mm3以上に回復しない場合は、1段階減量を維持する。 |
| 1.5万/mm3未満 | 2万/mm3以上になるまで休薬し、休薬前の用量注)から1段階減量して投与を再開する。 |
| 好中球数 | |
| 500/mm3以上750/mm3未満 | 1段階減量する。1,000/mm3超に回復した場合は、減量前の用量を再開する。 |
| 500/mm3未満 | 500/mm3を超えるまで休薬し、休薬前の用量注)から1段階減量して投与を再開する。1,000/mm3超に回復した場合は、休薬前の用量注)を再開してもよい。 |
| 総ビリルビン上昇:移植片対宿主病に伴う肝病変を有さない場合 | |
| 3×ULN超、5×ULN以下 | 3×ULN以下になるまで、1段階減量する。 |
| 5×ULN超、10×ULN以下 | 3×ULN以下になるまで最長14日間休薬する。14日以内に3×ULN以下に回復した場合は、休薬前の用量注)で投与を再開してもよい。14日を過ぎても3×ULN以下に回復しない場合は、休薬前の用量注)から1段階減量して投与を再開する。 |
| 10×ULN超 | 3×ULN以下になるまで休薬し、休薬前の用量注)から1段階減量して投与を再開する。 |
| 総ビリルビン上昇:移植片対宿主病に伴う肝病変を有する場合 | |
| 3×ULN超 | 3×ULN以下になるまで、1段階減量を継続する。 |
7.10 治療効果が認められた場合は、本剤の漸減を検討すること。本剤の漸減は、ステロイドの投与中止後に、2カ月ごとに1段階を目安とし、副作用により減量する場合の1段階減量と同じ減量幅とすること。なお、本剤の漸減中に症状が再発した場合は、本剤の漸増等の適切な対応を行うこと。
7.11 錠剤と液剤の生物学的同等性は示されていないため、可能な限り錠剤と液剤の切替えを避け、やむを得ず切り替える場合には、患者の状態を慎重に観察すること。
ジャカビ錠10mg
<骨髄線維症、真性多血症>
7.1 他の抗悪性腫瘍剤との併用について、有効性及び安全性は確立していない。
7.2 十分な効果が認められず、血球数から増量可能と判断できる場合は、1回の投与量を5mgずつ2週間以上の間隔をあけて増量することができる。ただし、本剤の初回投与後、4週間は増量しないこと。
<骨髄線維症>
7.3 本剤の投与開始にあたっては、血小板数に基づき次表を参考に開始用量を決定すること。
| 血小板数注) | 開始用量 |
| 20万/mm3超 | 1回20mg1日2回 |
| 10万/mm3以上20万/mm3以下 | 1回15mg1日2回 |
7.4 本剤の投与中に血小板数が減少した場合、下表を参考に減量又は休薬を考慮すること。なお、血小板数が休薬前の数値以上に回復した場合には、1回5mg1日2回から投与を再開できる。ただし、患者の状態をより慎重に観察し、有害事象の発現に十分注意すること。
| 血小板数 | 1回あたりの用量(1日2回) | ||||
| 25mg | 20mg | 15mg | 10mg | 5mg | |
| 10万/mm3以上12.5万/mm3未満 | 20mg | 変更なし | |||
| 7.5万/mm3以上10万/mm3未満 | 10mg | 10mg | 10mg | 変更なし | |
| 5万/mm3以上7.5万/mm3未満 | 5mg | 5mg | 5mg | 5mg | 変更なし |
| 5万/mm3未満 | 休薬 | ||||
7.5 本剤の投与中に好中球数が500/mm3未満に減少した場合には休薬すること。なお、好中球数が休薬前の数値以上に回復した場合には、1回5mg1日2回から投与を再開できる。ただし、患者の状態をより慎重に観察し、有害事象の発現に十分注意すること。
<真性多血症>
7.6 血小板数が5万/mm3以上10万/mm3未満の患者における開始用量の情報は得られていないため、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分理解した上で、本剤の投与の可否を慎重に検討すること。血小板数5万/mm3以上10万/mm3未満の患者に投与可能と判断する場合、低用量から投与を開始するとともに、観察を十分に行い、有害事象の発現に十分注意すること。血小板数5万/mm3未満の患者に対する投与は避けること。
7.7 本剤の投与中に血小板数又はヘモグロビンが減少した場合、下表を参考に減量又は休薬を考慮すること。減量幅は、1回の投与量として5mgとする。なお、血小板数及びヘモグロビンが休薬前の数値以上に回復した場合には、1回5mg1日2回から投与を再開できる。ただし、患者の状態をより慎重に観察し、有害事象の発現に十分注意すること。
| 血小板数 | 5万/mm3以上、10万/mm3未満 | 減量 |
| 5万/mm3未満 | 休薬 | |
| ヘモグロビン | 8g/dL以上、12g/dL未満 | 減量 |
| 8g/dL未満 | 休薬 |
7.8 本剤の投与中に好中球数が1,000/mm3未満に減少した場合には休薬すること。なお、好中球数が休薬前の数値以上に回復した場合には、1回5mg1日2回から投与を再開できる。ただし、患者の状態をより慎重に観察し、有害事象の発現に十分注意すること。
<造血幹細胞移植後の移植片対宿主病>
7.9 副作用により本剤を休薬、減量する場合は、以下の基準を考慮すること。[9.1.4参照]
| 投与量 | 減量又は休薬 |
| 1回10mg1日2回 | 1回5mg1日2回 |
| 1回5mg1日2回 | 1回5mg1日1回 |
| 1回5mg1日1回 | 休薬 |
| 1回4mg/m21日2回 | 1回2mg/m21日2回 |
| 1回2mg/m21日2回 | 休薬 |
| 血小板数 | |
| 1.5万/mm3以上2万/mm3未満 | 1段階減量する。減量後7日以内に2万/mm3以上に回復した場合は、減量前の用量を再開してもよい。 減量後7日を過ぎても2万/mm3以上に回復しない場合は、1段階減量を維持する。 |
| 1.5万/mm3未満 | 2万/mm3以上になるまで休薬し、休薬前の用量注)から1段階減量して投与を再開する。 |
| 好中球数 | |
| 500/mm3以上750/mm3未満 | 1段階減量する。1,000/mm3超に回復した場合は、減量前の用量を再開する。 |
| 500/mm3未満 | 500/mm3を超えるまで休薬し、休薬前の用量注)から1段階減量して投与を再開する。1,000/mm3超に回復した場合は、休薬前の用量注)を再開してもよい。 |
| 総ビリルビン上昇:移植片対宿主病に伴う肝病変を有さない場合 | |
| 3×ULN超、5×ULN以下 | 3×ULN以下になるまで、1段階減量する。 |
| 5×ULN超、10×ULN以下 | 3×ULN以下になるまで最長14日間休薬する。14日以内に3×ULN以下に回復した場合は、休薬前の用量注)で投与を再開してもよい。14日を過ぎても3×ULN以下に回復しない場合は、休薬前の用量注)から1段階減量して投与を再開する。 |
| 10×ULN超 | 3×ULN以下になるまで休薬し、休薬前の用量注)から1段階減量して投与を再開する。 |
| 総ビリルビン上昇:移植片対宿主病に伴う肝病変を有する場合 | |
| 3×ULN超 | 3×ULN以下になるまで、1段階減量を継続する。 |
7.10 治療効果が認められた場合は、本剤の漸減を検討すること。本剤の漸減は、ステロイドの投与中止後に、2カ月ごとに1段階を目安とし、副作用により減量する場合の1段階減量と同じ減量幅とすること。なお、本剤の漸減中に症状が再発した場合は、本剤の漸増等の適切な対応を行うこと。
7.11 錠剤と液剤の生物学的同等性は示されていないため、可能な限り錠剤と液剤の切替えを避け、やむを得ず切り替える場合には、患者の状態を慎重に観察すること。
ジャカビ内用液小児用0.5%
<造血幹細胞移植後の移植片対宿主病>
7.9 副作用により本剤を休薬、減量する場合は、以下の基準を考慮すること。[9.1.4参照]
| 投与量 | 減量又は休薬 |
| 1回10mg1日2回 | 1回5mg1日2回 |
| 1回5mg1日2回 | 1回5mg1日1回 |
| 1回5mg1日1回 | 休薬 |
| 1回4mg/m21日2回 | 1回2mg/m21日2回 |
| 1回2mg/m21日2回 | 休薬 |
| 血小板数 | |
| 1.5万/mm3以上2万/mm3未満 | 1段階減量する。減量後7日以内に2万/mm3以上に回復した場合は、減量前の用量を再開してもよい。 減量後7日を過ぎても2万/mm3以上に回復しない場合は、1段階減量を維持する。 |
| 1.5万/mm3未満 | 2万/mm3以上になるまで休薬し、休薬前の用量注)から1段階減量して投与を再開する。 |
| 好中球数 | |
| 500/mm3以上750/mm3未満 | 1段階減量する。1,000/mm3超に回復した場合は、減量前の用量を再開する。 |
| 500/mm3未満 | 500/mm3を超えるまで休薬し、休薬前の用量注)から1段階減量して投与を再開する。1,000/mm3超に回復した場合は、休薬前の用量注)を再開してもよい。 |
| 総ビリルビン上昇:移植片対宿主病に伴う肝病変を有さない場合 | |
| 3×ULN超、5×ULN以下 | 3×ULN以下になるまで、1段階減量する。 |
| 5×ULN超、10×ULN以下 | 3×ULN以下になるまで最長14日間休薬する。14日以内に3×ULN以下に回復した場合は、休薬前の用量注)で投与を再開してもよい。14日を過ぎても3×ULN以下に回復しない場合は、休薬前の用量注)から1段階減量して投与を再開する。 |
| 10×ULN超 | 3×ULN以下になるまで休薬し、休薬前の用量注)から1段階減量して投与を再開する。 |
| 総ビリルビン上昇:移植片対宿主病に伴う肝病変を有する場合 | |
| 3×ULN超 | 3×ULN以下になるまで、1段階減量を継続する。 |
7.10 治療効果が認められた場合は、本剤の漸減を検討すること。本剤の漸減は、ステロイドの投与中止後に、2カ月ごとに1段階を目安とし、副作用により減量する場合の1段階減量と同じ減量幅とすること。なお、本剤の漸減中に症状が再発した場合は、本剤の漸増等の適切な対応を行うこと。
7.11 錠剤と液剤の生物学的同等性は示されていないため、可能な限り錠剤と液剤の切替えを避け、やむを得ず切り替える場合には、患者の状態を慎重に観察すること。
8. 重要な基本的注意
8.1 血小板減少症、貧血、好中球減少症があらわれることがあるので、本剤の投与開始前及び投与中は、定期的に血液検査(血球数算定、白血球分画等)を行うこと。[11.1.1参照]
8.2 免疫抑制作用により、細菌、真菌、ウイルス又は原虫による感染症や日和見感染が発現又は悪化することがある。肝炎ウイルス、結核等が再活性化するおそれがあるので、本剤投与に先立って肝炎ウイルス、結核等の感染の有無を確認し、本剤の投与開始前に適切な処置の実施を考慮すること。本剤投与中は感染症の発現又は増悪に十分注意すること。[1.2、9.1.1-9.1.3、11.1.2参照]
8.3 帯状疱疹があらわれることがあるので、本剤の投与開始前に、患者に対して帯状疱疹の初期症状について説明し、異常が認められた場合には速やかに連絡し、適切な処置を受けるよう指導すること。[11.1.2参照]
8.4 出血があらわれることがあるので、本剤投与中は定期的に血液検査等を実施すること。[11.1.4参照]
8.5 肝機能障害があらわれることがあるので、本剤投与中は定期的に肝機能検査等を実施すること。[11.1.6参照]
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 結核の既感染者(特に結核の既往歴のある患者及び胸部レントゲン上結核治癒所見のある患者)
9.1.2 感染症(敗血症、肺炎、ウイルス感染等)を合併している患者
9.1.3 B型肝炎ウイルスキャリアの患者又はHBs抗原陰性かつHBc抗体若しくはHBs抗体陽性の患者
9.1.4 移植片対宿主病に伴う肝病変(総ビリルビン値が正常値上限の3倍以上)を有する患者
より頻回に血球数を測定し、投与量を調節することが望ましい。[7.9参照]
9.2 腎機能障害患者
減量を考慮するとともに、患者の状態をより慎重に観察し、有害事象の発現に十分注意すること。活性代謝物の血中濃度が上昇するとの報告がある。[16.6.1参照]
9.3 肝機能障害患者
減量を考慮するとともに、患者の状態をより慎重に観察し、有害事象の発現に十分注意すること。未変化体の血中濃度が上昇するとの報告がある。[16.6.2参照]
9.4 生殖能を有する者
妊娠する可能性のある女性には、本剤投与中において避妊する必要性及び適切な避妊法について説明すること。[9.5参照]
9.5 妊婦
9.6 授乳婦
授乳しないことが望ましい。動物実験(ラット)において、本剤及び本剤の代謝物が乳汁中に移行し、母体血漿中濃度の13倍であったとの報告がある。
9.7 小児等
<骨髄線維症、真性多血症>
小児等を対象とした臨床試験は実施していない。
<造血幹細胞移植後の移植片対宿主病>
28日齢未満の小児等を対象とした臨床試験は実施していない。また、2歳未満の患者に対する本剤の用法及び用量の適切性について、臨床試験で十分な検討は行われていない。[16.6.3参照]
9.8 高齢者
患者の状態を十分に観察し、慎重に投与すること。臨床試験において、高齢者(65歳超)では、65歳以下の患者と比較して、血小板減少症、心不全等の発現が増加することが報告されている。
10. 相互作用
相互作用序文
本剤は主として代謝酵素CYP3A4で代謝され、CYP3A4に比べて寄与率は小さいがCYP2C9によっても代謝される。また、in vitroの検討から、本剤はP-糖蛋白(P-gp)及び乳癌耐性蛋白(BCRP)を阻害する可能性が示唆されている。
薬物代謝酵素用語
CYP3A4
薬物代謝酵素用語
CYP2C9
薬物代謝酵素用語
P-糖蛋白(P-gp)
薬物代謝酵素用語
乳癌耐性蛋白(BCRP)
10.2 併用注意
| 強力なCYP3A4阻害剤 イトラコナゾール リトナビル クラリスロマイシン等 [16.7.1参照] | 本剤の血中濃度が上昇するおそれがあるので、CYP3A4阻害作用のない又は弱い薬剤への代替を考慮すること。やむを得ず強力なCYP3A4阻害剤と本剤を併用投与する場合には、本剤の減量を考慮するとともに、患者の状態を慎重に観察し、有害事象の発現に十分注意すること。 | これらの薬剤の強力なCYP3A4阻害作用により、本剤の代謝が阻害されると考えられる。 |
| CYP3A4及びCYP2C9を阻害する薬剤 フルコナゾール等 | 本剤の血中濃度が上昇するおそれがある。 | これらの薬剤の2つの代謝酵素(CYP3A4及びCYP2C9)の阻害作用により、本剤の代謝が阻害されると考えられる。 |
| CYP3A4阻害剤 エリスロマイシン シプロフロキサシン アタザナビル ジルチアゼム シメチジン等 [16.7.2参照] | 本剤の血中濃度が上昇するおそれがあるので、CYP3A4阻害剤と本剤を併用投与する場合には、患者の状態を慎重に観察し、有害事象の発現に十分注意すること。 | これらの薬剤のCYP3A4阻害作用により、本剤の代謝が阻害されると考えられる。 |
| CYP3A4誘導剤 リファンピシン フェニトイン セイヨウオトギリソウ〔St.John's Wort(セント・ジョーンズ・ワート)〕含有食品等 [16.7.3参照] | 本剤の血中濃度が低下し、本剤の有効性が減弱する可能性があるので、CYP3A4誘導作用のない又は弱い薬剤への代替を考慮すること。 | これらの薬剤のCYP3A4誘導作用により、本剤の代謝が促進されると考えられる。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 骨髄抑制
血小板減少症(33.3%)、貧血(29.9%)、好中球減少症(10.7%)、汎血球減少症(0.9%)等があらわれることがある。[8.1参照]
11.1.2 感染症(16.7%)
11.1.3 進行性多巣性白質脳症(PML)(頻度不明)
本剤投与中及び投与終了後は患者の状態を十分に観察し、意識障害、認知障害、麻痺症状(片麻痺、四肢麻痺)、言語障害等の症状があらわれた場合には、MRIによる画像診断及び脳脊髄液検査を実施するとともに、投与を中止し、適切な処置を行うこと。
11.1.4 出血
脳出血等の頭蓋内出血(0.1%)(初期症状:頭痛、悪心・嘔吐、意識障害、片麻痺等)、胃腸出血(1.2%)、処置後出血(0.1%)、鼻出血(1.3%)、血尿(0.7%)等があらわれることがあり、死亡に至った症例が報告されている。[8.4参照]
11.1.5 間質性肺疾患(頻度不明)
11.1.6 肝機能障害
AST(3.2%)、ALT(4.9%)の上昇等を伴う肝機能障害があらわれることがあり、死亡に至った症例が報告されている。[8.5参照]
11.1.7 心不全(0.4%)
11.2 その他の副作用
| 5%以上 | 1〜5%未満 | 1%未満 | |
| 感染症 | − | 肺炎、敗血症、上咽頭炎 | サイトメガロウイルス感染、BKウイルス感染 |
| 血液及びリンパ系障害 | 白血球数減少 | − | − |
| 代謝及び栄養障害 | − | 体重増加、高コレステロール血症 | 高トリグリセリド血症、体液貯留、低カルシウム血症、食欲減退 |
| 精神障害 | − | 不眠症 | − |
| 神経系障害 | − | 頭痛、浮動性めまい | 末梢性ニューロパチー、錯感覚 |
| 心臓障害 | − | − | 動悸 |
| 血管障害 | − | 高血圧 | − |
| 呼吸器系障害 | − | 呼吸困難、咳嗽 | ラ音 |
| 胃腸障害 | 下痢 | 悪心、腹痛、嘔吐、便秘、腹部膨満、口内炎、鼓腸、上腹部痛 | 口内乾燥、口腔内潰瘍形成、消化不良、リパーゼ上昇、アミラーゼ上昇 |
| 肝胆道系障害 | − | γ-GTP増加、ALP増加、血中ビリルビン増加 | − |
| 皮膚及び皮下組織障害 | − | 挫傷 | 発疹、寝汗 |
| 筋骨格系障害 | − | 筋痙縮、四肢痛、筋肉痛、関節痛、血中CK上昇 | 骨痛、背部痛 |
| 腎及び尿路障害 | − | 血中尿素増加、血中クレアチニン上昇 | − |
| 全身障害 | − | 末梢性浮腫、無力症、発熱、疲労 | − |
| 臨床検査 | − | − | APTT延長 |
14. 適用上の注意
<錠5mg・10mg>
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
<内用液>
14.2 薬剤調製時の注意
体表面積あたりの用量で使用する際には巻末の投与液量一覧表を参考に調製すること。
15. その他の注意
15.1 臨床使用に基づく情報
15.1.1 心血管系事象のリスク因子を有する関節リウマチ患者を対象としたJAK阻害剤トファシチニブクエン酸塩の海外臨床試験の結果、主要評価項目である主要な心血管系事象(Major Adverse Cardiovascular Events:MACE)及び悪性腫瘍(非黒色腫皮膚癌を除く)の発現率について、TNF阻害剤群に対するハザード比(95%信頼区間)はそれぞれ1.33(0.91,1.94)及び1.48(1.04,2.09)であり、95%信頼区間上限は予め設定していた非劣性マージン1.8を超え、TNF阻害剤群に対する非劣性が検証されなかったことが報告されている。また、本剤でも、国内市販後の自発報告において、心血管系事象の発現が認められている。
15.2 非臨床試験に基づく情報
15.2.1 イヌを用いた心血管系への影響に関する試験では、心拍数増加を伴う血圧低下が認められ、ラットを用いた呼吸機能検査では、分時換気量減少が認められた。
15.2.2 イヌを用いた26及び52週間反復投与毒性試験において、皮膚乳頭腫の発現が認められた。また、本剤との因果関係は明らかでないものの、本剤投与後に非黒色腫皮膚癌(基底細胞癌、扁平上皮癌、メルケル細胞癌を含む)等の悪性腫瘍(二次発がん)の発現が報告されている。
15.2.3 幼若ラットを用いた毒性試験において、骨成長の抑制と骨折が認められた。幼若ラットでの曝露量(AUC)は、最大推奨用量を投与した成人でのAUCの1.5倍(骨成長の抑制)、13倍(骨折)であった。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
健康被験者にルキソリチニブ10、25、50及び100mgを空腹時に単回経口投与したとき、未変化体の血漿中濃度は投与後0.5時間(Tmax中央値)でCmaxに達し、その後、2.5〜3.4時間の半減期で消失した。Cmax及びAUCは投与量にほぼ比例した1)。
注)本剤の承認された用法及び用量での1日最大用量は50mgである。
| 投与量 | Cmax(nmol/L) | Tmax注)(h) | T1/2(h) | AUCinf(h・nmol/L) | CL/F(L/h) |
| 10mg(n=8) | 621±107(613) | 0.5(0.25-1.5) | 3.18±1.31(2.98) | 2,290±914(2,160) | 15.9±4.89(15.1) |
| 25mg(n=8) | 1,450±718(1,320) | 0.5(0.25-1.5) | 2.51±0.638(2.44) | 4,020±1,220(3,830) | 22.6±9.09(21.3) |
| 50mg(n=8) | 2,380±495(2,330) | 0.5(0.25-1.5) | 2.86±0.542(2.81) | 8,650±2,230(8,430) | 19.8±4.20(19.4) |
| 100mg(n=8) | 5,430±1,260(5,300) | 0.5(0.25-1.5) | 3.40±0.907(3.28) | 22,600±7,780(21,500) | 15.9±4.94(15.2) |
健康被験者にルキソリチニブ10、25、50及び100mgを単回経口投与したときの血漿中濃度推移(平均値±標準偏差)
16.1.2 反復投与
健康被験者にルキソリチニブ10及び25mgを7日間1日2回反復経口投与したときAUCの累積比はそれぞれ1.12及び1.03で大きな累積は認められなかった1)。
| 投与量 | 反復投与 | Cmax(nmol/L) | Tmax注)(h) | AUC0-12h(h・nmol/L) | AUC0-12h比(7日目/初日) |
| 10mg(n=8) | 1日目 | 577±70.8(573) | 0.375(0.25-1.0) | 1,920±678(1,830) | − |
| 7日目 | 587±187(562) | 0.5(0.25-1.0) | 2,180±949(2,040) | 1.12±0.117(1.11) | |
| 25mg(n=8) | 1日目 | 1,200±357(1,160) | 0.5(0.25-1.5) | 3,600±838(3,500) | − |
| 7日目 | 1,290±271(1,260) | 0.5(0.25-0.5) | 3,720±864(3,620) | 1.03±0.0568(1.03) |
16.2 吸収
16.2.1 食事の影響
健康被験者(16例)に食後にルキソリチニブ20mgを単回経口投与したとき、空腹時に比べTmaxは0.5時間から1.75時間に延長し、Cmaxは42%低下した。AUCは6.4%低下したが比(食後/空腹)の90%信頼区間は0.80〜1.25の範囲内であった2)。
16.3 分布
ルキソリチニブのヒト血漿中及び血清中での非結合型分率は、3.2〜4.8%であった3)(in vitro)。
16.4 代謝
ルキソリチニブは主としてCYP3A4で代謝され、またCYP3A4に比べて寄与率は小さいがCYP2C9によっても代謝されると考えられる4)(in vitro)。
16.5 排泄
健康被験者(6例)に14C標識したルキソリチニブ25mgを単回経口投与したとき放射能の総回収率は96%で、尿及び糞中にそれぞれ74%及び22%が回収された。尿及び糞中に回収された放射能に占める未変化体の割合は1%未満であった。放射能の70%以上が投与後24時間以内に回収された5)(外国人のデータ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
健康被験者(クレアチニンクリアランス(CLcr)80mL/min超)、軽度腎機能障害患者(CLcr50〜80mL/min)、中等度腎機能障害患者(CLcr30〜49mL/min)、重度腎機能障害患者(CLcr30mL/min未満)及び透析を受けている末期腎機能障害患者にルキソリチニブ25mgを単回経口投与したとき、未変化体の血漿中濃度は同様であった(各群8例)。8種類の活性代謝物のAUC(合計)は、未変化体のAUCに対して、健康被験者で61%、軽度、中等度及び重度腎機能障害患者で79%、117%及び173%、投与前及び投与後に透析を行った患者で346%及び297%で、腎機能障害の重症度の上昇により増加する傾向を示した6)(外国人のデータ)。[9.2参照]
16.6.2 肝機能障害患者
16.6.3 小児等
造血幹細胞移植後の28日齢以上18歳未満の急性移植片対宿主病(GVHD)患者を対象とした非盲検単群試験において、2歳以上6歳未満、6歳以上12歳未満及び12歳以上18歳未満の患者にそれぞれ開始用量として内用液4mg/m2、錠剤5mg及び錠剤10mgを1日2回とし経口投与したとき初回投与後における未変化体の血漿中濃度は投与後1.0〜1.5時間(Tmax中央値)でCmaxに達し、その後、1.36〜2.05時間の半減期で消失した8)。[9.7参照]
| 年齢[製剤] | 投与量 | Cmax(ng/mL) | Tmax注)(h) | T1/2(h) | AUC0-12h(h・ng/mL) |
| 2歳以上6歳未満[内用液] | 4mg/m2(n=8) | 77.6±52.1(66.5) | 1.00(0.500-4.00) | 2.05±1.19a)(1.78) | 336±261a)(282) |
| 6歳以上12歳未満[錠剤] | 5mg(n=8) | 124±74.4(105) | 1.50(0.500-8.03) | 1.68±0.294a)(1.66) | 461±170a)(438) |
| 12歳以上18歳未満[錠剤] | 10mg(n=5) | 96.3±68.5(66.1) | 1.50(1.00-8.97) | 1.36b)(1.07,1.65) | 248b)(128,367) |
16.7 薬物相互作用
16.7.1 ケトコナゾール(強力なCYP3A4阻害剤、国内未発売の経口剤)
16.7.2 エリスロマイシン(CYP3A4阻害剤)
16.7.3 リファンピシン(CYP3A4誘導剤)
16.7.4 ルキソリチニブの経口投与後、腸で薬物濃度が高くなった場合、P-糖蛋白(P-gp)及び乳癌耐性蛋白(BCRP)を阻害する可能性が示唆された10)(in vitro)。
16.7.5 ミダゾラム(CYP3A4基質)
健康被験者(23例)にルキソリチニブ25mg(1日2回1日間)を反復投与時、ミダゾラム経口液剤4mgを併用したとき、ルキソリチニブはミダゾラムの薬物動態に大きな影響を及ぼさなかった11)(外国人のデータ)。
16.7.6 経口避妊薬
健康被験者(24例)にルキソリチニブ25mg(1日2回10日間)を反復投与時、経口避妊薬(エチニルエストラジオール30μg及びレボノルゲストレル150μgを含有)を併用したとき、ルキソリチニブはエチニルエストラジオール及びレボノルゲストレルの薬物動態に影響を及ぼさなかった12)(外国人のデータ)。
17. 臨床成績
17.1 有効性及び安全性に関する試験
<骨髄線維症>
17.1.1 国際共同第II相試験(A2202試験)
骨髄線維症患者※1を対象とした非盲検非対照試験において、ベースラインの血小板数に基づき本剤を経口投与した。本剤の開始用量は、ベースラインの血小板数が10万〜20万/mm3の場合15mg1日2回、20万/mm3超の場合20mg1日2回とした。
合計120例(日本人患者30例を含む)に本剤が投与された。骨髄線維症患者における合併症の主な要因13)である脾腫に関して、主要評価項目である24週時に脾臓容積がベースラインから35%以上縮小した被験者の割合は31.7%であった14)。
副作用発現頻度は、本剤投与群で92.5%(111/120例(日本人30例を含む))であった。主な副作用は、貧血58.3%(70/120例)、血小板数減少28.3%(34/120例)、血小板減少症26.7%(32/120例)等であった。
(2013年6月7日カットオフ)
合計120例(日本人患者30例を含む)に本剤が投与された。骨髄線維症患者における合併症の主な要因13)である脾腫に関して、主要評価項目である24週時に脾臓容積がベースラインから35%以上縮小した被験者の割合は31.7%であった14)。
副作用発現頻度は、本剤投与群で92.5%(111/120例(日本人30例を含む))であった。主な副作用は、貧血58.3%(70/120例)、血小板数減少28.3%(34/120例)、血小板減少症26.7%(32/120例)等であった。
(2013年6月7日カットオフ)
17.1.2 海外第III相試験(351試験)
骨髄線維症患者※1を対象とした二重盲検無作為化比較試験において、ベースラインの血小板数に基づき本剤を経口投与した。本剤の開始用量は、ベースラインの血小板数が10万〜20万/mm3の場合15mg1日2回、20万/mm3超の場合20mg1日2回とした。
合計309例がルキソリチニブ群(155例)又はプラセボ群(154例)に無作為に割付けされた。主要評価項目である24週時に脾臓容積がベースラインから35%以上縮小した被験者の割合はルキソリチニブ群で41.9%、プラセボ群で0.7%であり、プラセボ群と比較してルキソリチニブ群で有意に高かった15)(Fisherの正確検定p<0.0001)。
副作用発現頻度は、本剤投与群で76.1%(118/155例)であった。主な副作用は、本剤投与群では血小板減少症34.2%(53/155例)、貧血25.2%(39/155例)、疲労12.9%(20/155例)等であった。
(2013年1月25日カットオフ)
合計309例がルキソリチニブ群(155例)又はプラセボ群(154例)に無作為に割付けされた。主要評価項目である24週時に脾臓容積がベースラインから35%以上縮小した被験者の割合はルキソリチニブ群で41.9%、プラセボ群で0.7%であり、プラセボ群と比較してルキソリチニブ群で有意に高かった15)(Fisherの正確検定p<0.0001)。
副作用発現頻度は、本剤投与群で76.1%(118/155例)であった。主な副作用は、本剤投与群では血小板減少症34.2%(53/155例)、貧血25.2%(39/155例)、疲労12.9%(20/155例)等であった。
(2013年1月25日カットオフ)
17.1.3 海外第III相試験(A2352試験)
骨髄線維症患者※1を対象とした非盲検無作為化比較試験において、ベースラインの血小板数に基づき本剤を経口投与した。本剤の開始用量は、ベースラインの血小板数が10万〜20万/mm3の場合15mg1日2回、20万/mm3超の場合20mg1日2回とした。
合計219例がルキソリチニブ群(146例)又はBest Available Therapy群(73例)に無作為に割付けされた。主要評価項目である48週時に脾臓容積がベースラインから35%以上縮小した被験者の割合はルキソリチニブ群で28.5%、Best Available Therapy群で0%であり、Best Available Therapy群と比較してルキソリチニブ群で有意に高かった16)(p<0.0001、Cochran-Mantel-Haenszelの正確検定)。
副作用発現頻度は、本剤投与群で82.9%(121/146例)であった。主な副作用は、本剤投与群では血小板減少症43.8%(64/146例)、貧血32.9%(48/146例)、体重増加11.0%(16/146例)等であった。
(2012年12月1日カットオフ)
合計219例がルキソリチニブ群(146例)又はBest Available Therapy群(73例)に無作為に割付けされた。主要評価項目である48週時に脾臓容積がベースラインから35%以上縮小した被験者の割合はルキソリチニブ群で28.5%、Best Available Therapy群で0%であり、Best Available Therapy群と比較してルキソリチニブ群で有意に高かった16)(p<0.0001、Cochran-Mantel-Haenszelの正確検定)。
副作用発現頻度は、本剤投与群で82.9%(121/146例)であった。主な副作用は、本剤投与群では血小板減少症43.8%(64/146例)、貧血32.9%(48/146例)、体重増加11.0%(16/146例)等であった。
(2012年12月1日カットオフ)
※1:試験対象患者
<真性多血症>
17.1.4 国際共同第III相試験(B2301試験)
真性多血症患者※2を対象とした非盲検無作為化比較試験において、開始用量10mg1日2回とし、被験者の状態により5mg1日1回から25mg1日2回の範囲で本剤を経口投与した。合計222例(日本人患者18例を含む)がルキソリチニブ群(110例)又はBest Available Therapy群(112例)に無作為に割付けされた。主要評価項目である32週時の奏効※3率はルキソリチニブ群で22.7%、Best Available Therapy群で0.9%であり、Best Available Therapy群と比較してルキソリチニブ群で有意に高かった20)(p<0.0001、層別Cochran-Mantel-Haenszel検定)。
48週時点での副作用発現頻度は、本剤投与群で70.9%(78/110例(日本人6例を含む))であった。主な副作用は、貧血21.8%(24/110例)、血小板減少症10.9%(12/110例)、体重増加8.2%(9/110例)等であった。
48週時点での副作用発現頻度は、本剤投与群で70.9%(78/110例(日本人6例を含む))であった。主な副作用は、貧血21.8%(24/110例)、血小板減少症10.9%(12/110例)、体重増加8.2%(9/110例)等であった。
※2:試験対象患者
※3:奏効は、以下の両基準に該当した場合
・ヘマトクリットコントロール
瀉血実施基準を「連続2回の検査で、ヘマトクリット値が45%超かつベースライン値より3%以上高い、又は48%超のいずれかに該当する場合」とし、無作為化から8週時まで瀉血実施1回以下、かつ8週時から32週時まで瀉血実施不要。
・脾臓容積35%以上縮小
32週時のMRI又はCTに基づく脾臓容積がベースラインから35%以上縮小。
<造血幹細胞移植後の移植片対宿主病>
17.1.5 国際共同第III相試験(C2301試験)
造血幹細胞移植後の急性移植片対宿主病(GVHD)患者※4を対象とした非盲検無作為化比較試験において、開始用量10mg1日2回とし本剤を経口投与した。合計309例(日本人患者30例を含む)がルキソリチニブ群(154例)又はBest Available Therapy群(155例)に無作為に割付けされた。主要評価項目である投与28日時の奏効※5率はルキソリチニブ群で62.3%、Best Available Therapy群で39.4%であり、Best Available Therapy群と比較してルキソリチニブ群で有意に高かった22)(p<0.0001、Cochran-Mantel-Haenszel検定)。
副作用発現頻度は、本剤投与群で66.4%(101/152例(日本人9例を含む))であった。主な副作用は、血小板減少症23.0%(35/152例)、貧血16.4%(25/152例)、血小板数減少14.5%(22/152例)等であった。
(2020年1月6日カットオフ)
副作用発現頻度は、本剤投与群で66.4%(101/152例(日本人9例を含む))であった。主な副作用は、血小板減少症23.0%(35/152例)、貧血16.4%(25/152例)、血小板数減少14.5%(22/152例)等であった。
(2020年1月6日カットオフ)
※4:12歳以上のステロイド抵抗性急性GVHD(グレードII〜IV)の患者
※5:追加の全身治療がなく、国際標準基準23)で完全奏効又は部分奏効を得られた患者
17.1.6 国際共同第III相試験(D2301試験)
造血幹細胞移植後の慢性GVHD患者※6を対象とした非盲検無作為化比較試験において、開始用量10mg1日2回とし本剤を経口投与した。合計329例(日本人患者37例を含む)がルキソリチニブ群(165例)又はBest Available Therapy群(164例)に無作為に割付けされた。中間解析を行い、主要評価項目及び2つの主な副次的評価項目のいずれかで統計学的有意差が認められなかった場合には試験を続行する計画であった。中間解析(2019年7月9日カットオフ)において、ルキソリチニブ群97例、Best Available Therapy群99例が評価され、主要評価項目である投与24週時の奏効※7率はルキソリチニブ群で50.5%、Best Available Therapy群で26.3%であり、Best Available Therapy群と比較してルキソリチニブ群で有意に高かった24)(p=0.0003、Cochran-Mantel-Haenszel検定、有意水準片側1.176%)。
最終解析(2020年5月8日カットオフ)において、主要評価項目である投与24週時の奏効※7率はルキソリチニブ群で49.7%、Best Available Therapy群で25.6%であり、Best Available Therapy群と比較してルキソリチニブ群で高かった24)。
副作用発現頻度は、本剤投与群で67.9%(112/165例(日本人22例を含む))であった。主な副作用は、貧血23.6%(39/165例)、好中球減少症10.9%(18/165例)、ALT増加10.3%(17/165例)等であった。
最終解析(2020年5月8日カットオフ)において、主要評価項目である投与24週時の奏効※7率はルキソリチニブ群で49.7%、Best Available Therapy群で25.6%であり、Best Available Therapy群と比較してルキソリチニブ群で高かった24)。
副作用発現頻度は、本剤投与群で67.9%(112/165例(日本人22例を含む))であった。主な副作用は、貧血23.6%(39/165例)、好中球減少症10.9%(18/165例)、ALT増加10.3%(17/165例)等であった。
※6:12歳以上のステロイド抵抗性慢性GVHD(中等症又は重症)の患者
※7:追加の全身治療がなく、NIH基準25)で完全奏効又は部分奏効を得られた患者
17.1.7 国際共同第I/II相試験(F12201試験)
造血幹細胞移植後の小児急性GVHD患者※8を対象とした非盲検単群試験において、ルキソリチニブの錠剤、カプセル剤※9又は液剤を年齢グループ別の開始用量(12歳以上18歳未満では10mg、6歳以上12歳未満では5mg、2歳以上6歳未満では4mg/m2)で1日2回経口投与した。合計45例(日本人患者6例を含む)にルキソリチニブが投与された。主要評価項目である投与28日時の奏効※10率は84.4%(90%CI:72.8%,92.5%)であった8)。なお、ステロイド抵抗性の患者の投与28日時の奏効率は90.6%(90%CI:77.5%,97.4%(29/32例)であった。
副作用発現頻度は、51.1%(23/45例)であった。主な副作用は、貧血20.0%(9/45例)、好中球数減少17.8%(8/45例)、白血球数減少15.6%(7/45例)等であった。
(2023年2月2日カットオフ)
副作用発現頻度は、51.1%(23/45例)であった。主な副作用は、貧血20.0%(9/45例)、好中球数減少17.8%(8/45例)、白血球数減少15.6%(7/45例)等であった。
(2023年2月2日カットオフ)
※8:28日齢以上18歳未満の未治療又はステロイド抵抗性急性GVHD(グレードII〜IV)の患者。なお、本剤の承認された効能又は効果は「造血幹細胞移植後の移植片対宿主病(ステロイド剤の投与で効果不十分な場合)」である。
※9:国内未承認
※10:追加の全身治療がなく、国際標準基準23)で完全奏効又は部分奏効を得られた患者
17.1.8 国際共同第II相試験(G12201試験)
造血幹細胞移植後の小児慢性GVHD患者※11を対象とした非盲検単群試験において、ルキソリチニブの錠剤又は液剤を年齢グループ別の開始用量(12歳以上18歳未満では10mg、6歳以上12歳未満では5mg、2歳以上6歳未満では4mg/m2)で1日2回経口投与した。合計45例(日本人患者7例を含む)にルキソリチニブが投与された。中間解析(2022年10月19日カットオフ)において、主要評価項目である投与24週時の奏効※12率は40.0%(90%CI:27.7%,53.3%)であった26)。なお、ステロイド抵抗性の患者の投与24週時の奏効率は39.3%(90%CI:23.8%,56.5%)(11/28例)であった。
副作用発現頻度は、57.8%(26/45例)であった。主な副作用は、貧血15.6%(7/45例)、好中球減少症13.3%(6/45例)、好中球数減少11.1%(5/45例)、血小板数減少11.1%(5/45例)等であった。
副作用発現頻度は、57.8%(26/45例)であった。主な副作用は、貧血15.6%(7/45例)、好中球減少症13.3%(6/45例)、好中球数減少11.1%(5/45例)、血小板数減少11.1%(5/45例)等であった。
※11:28日齢以上18歳未満の未治療又はステロイド抵抗性慢性GVHD(中等症又は重症)の患者。なお、本剤の承認された効能又は効果は「造血幹細胞移植後の移植片対宿主病(ステロイド剤の投与で効果不十分な場合)」である。
※12:追加の全身治療がなく、NIH基準25)で完全奏効又は部分奏効を得られた患者
18. 薬効薬理
18.1 作用機序
ルキソリチニブは、JAK1及びJAK2を選択的に阻害し、STAT等を介したサイトカイン及び成長因子のシグナル伝達を抑制することで、造血及び免疫機能を制御する。
18.2 薬理作用
18.2.1 JAK1及びJAK2阻害作用(in vitro)
18.2.2 動物モデルにおける造血系腫瘍抑制作用(in vivo)
18.2.3 動物モデルにおけるGVHD抑制作用(in vivo)
19. 有効成分に関する理化学的知見
19.1. ルキソリチニブリン酸塩
| 一般的名称 | ルキソリチニブリン酸塩 |
|---|---|
| 一般的名称(欧名) | Ruxolitinib Phosphate |
| 化学名 | (3R)-3-Cyclopentyl-3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]propanenitrile monophosphate |
| 分子式 | C17H18N6・H3PO4 |
| 分子量 | 404.36 |
| 融点 | 194〜198℃ |
| 物理化学的性状 | 白色の粉末である。水にやや溶けやすく、エタノールにやや溶けにくく、アセトニトリルに極めて溶けにくい。 |
| 分配係数 | −0.057(1-オクタノール/pH1.0緩衝液)、2.562(1-オクタノール/pH4.3緩衝液)、2.814(1-オクタノール/pH7.4緩衝液) |
| KEGG DRUG |
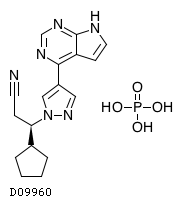
|
20. 取扱い上の注意
<内用液>
開封後60日以内に使用すること。
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
<ジャカビ錠5mg>
20錠[10錠(PTP)×2]
120錠[10錠(PTP)×12]
<ジャカビ錠10mg>
20錠[10錠(PTP)×2]
<ジャカビ内用液小児用0.5%>
60mL[1瓶]
23. 主要文献
- 社内資料:国内第I相臨床試験(1101試験)(2014年7月4日承認、CTD2.7.2-2.2.1)
- 社内資料:国内第I相臨床試験(1102試験)(2014年7月4日承認、CTD2.7.2-2.2.2)
- 社内資料:蛋白結合率(2014年7月4日承認、CTD2.7.2-2.1.1)
- 社内資料:CYP代謝酵素の同定(2014年7月4日承認、CTD2.7.2-2.1.2)
- Shilling,A.D.et al., Drug Metab.Dispos., 38 (11), 2023-2031, (2010) »PubMed
- 社内資料:腎機能障害患者を対象とした試験(142試験)(2014年7月4日承認、CTD2.7.2-2.4.2)
- 社内資料:肝機能障害患者を対象とした試験(137試験)(2014年7月4日承認、CTD2.7.2-2.4.1)
- 社内資料:国際共同第I/II相試験(F12201試験)
- Shi.J.G.,Chen.X.,Emm.T.et al., J.Clin.Pharmacol., 52 (6), 809-818, (2012) »PubMed
- 社内資料:膜透過性,薬物トランスポーター阻害及び腸でのCYP3A4,Pgp,BCRP阻害(2014年7月4日承認、CTD2.7.2-2.1.3)
- 社内資料:ミダゾラムとの薬物相互作用(2103試験)(2015年9月24日承認、CTD2.7.2-2.2)
- 社内資料:経口避妊薬(エチニルエストラジオール及びレボノルゲストレル)との薬物相互作用(2102試験)(2015年9月24日承認、CTD2.7.2-2.1)
- Mesa,R.A., Blood, 113 (22), 5394-5400, (2009) »PubMed
- 社内資料:骨髄線維症患者を対象としたアジア国際共同第II相臨床試験(2202試験)(2014年7月4日承認、CTD2.7.6-4.2.1)
- 社内資料:骨髄線維症患者を対象とした海外第III相臨床試験(351試験)(2014年7月4日承認、CTD2.7.6-4.1.1)
- 社内資料:骨髄線維症患者を対象とした海外第III相臨床試験(2352試験)(2014年7月4日承認、CTD2.7.6-4.1.2)
- Tefferi,A.and Vardiman,J.W., Leukemia, 22 (1), 14-22, (2008) »PubMed
- Barosi,G.et al., Leukemia, 22 (2), 437-438, (2008) »PubMed
- Cervantes,F.et al., Blood, 113 (13), 2895-2901, (2009) »PubMed
- 社内資料:真性多血症患者を対象とした国際共同第III相臨床試験(2301試験)(2015年9月24日承認、CTD2.7.6-4.1.1)
- Barosi,G.et al., Br.J.Haematol., 148 (6), 961-963, (2010) »PubMed
- 社内資料:急性移植片対宿主病を対象とした国際共同第III相臨床試験(C2301試験)(2023年8月23日承認、CTD2.7.6-4.1.1)
- Harris AC,Young R,Devine S,et al., Biol Blood Marrow Transplant, 22 (1), 4-10, (2016) »PubMed
- 社内資料:慢性移植片対宿主病を対象とした国際共同第III相臨床試験(D2301試験)(2023年8月23日承認、CTD2.7.6-4.1.2)
- Lee SJ,Wolff D,Kitko C,et al., Biol Blood Marrow Transplant, 21 (6), 984-999, (2015) »PubMed
- 社内資料:国際共同第II相試験(G12201試験)
- 社内資料:In vitro酵素阻害作用(2014年7月4日承認、CTD2.6.2-2.1)
- 社内資料:In vitro腫瘍増殖抑制作用(2014年7月4日承認、CTD2.6.2-2.2)
- 社内資料:In vivo腫瘍増殖抑制作用(2014年7月4日承認、CTD2.6.2-2.3)
- 社内資料:In vivoサイトカイン産生抑制作用(2014年7月4日承認、CTD2.6.2-2.3)
- 社内資料:In vivo(変異型JAK)腫瘍増殖抑制作用(2015年9月24日承認、CTD2.6.2-2.2.1)
- 社内資料:In vivo急性GVHD抑制作用(2023年8月23日承認、CTD2.6.2-2.1)
- 社内資料:In vivo慢性GVHD抑制作用(2023年8月23日承認、CTD2.6.2-2.2)
24. 文献請求先及び問い合わせ先
文献請求先
ノバルティスファーマ株式会社
ノバルティスダイレクト
〒105-6333
東京都港区虎ノ門1-23-1
電話:0120-003-293 受付時間:月〜金 9:00〜17:30(祝日及び当社休日を除く)
製品情報問い合わせ先
ノバルティスファーマ株式会社
ノバルティスダイレクト
〒105-6333
東京都港区虎ノ門1-23-1
電話:0120-003-293 受付時間:月〜金 9:00〜17:30(祝日及び当社休日を除く)
26. 製造販売業者等
26.1 製造販売(輸入)
ノバルティスファーマ株式会社
東京都港区虎ノ門1-23-1
その他の説明
その他の説明
<参考>
ジャカビ内用液小児用0.5%投与液量一覧表
造血幹細胞移植後の移植片対宿主病(ステロイド剤の投与で効果不十分な場合)
6歳未満の小児患者に対する投与液量=用量レベル×患者の体表面積÷投与濃度
| 体表面積 | ルキソリチニブ投与液量(1回あたり) | 投与濃度 | |
| 最小 | 最大 | ||
| 0.07m2 | 0.18m2 | 0.1mL | 5mg/mL |
| 0.19m2 | 0.31m2 | 0.2mL | |
| 0.32m2 | 0.43m2 | 0.3mL | |
| 0.44m2 | 0.56m2 | 0.4mL | |
| 0.57m2 | 0.68m2 | 0.5mL | |
| 0.69m2 | 0.81m2 | 0.6mL | |
| 0.82m2 | 0.93m2 | 0.7mL | |
| 0.94m2 | 1.06m2 | 0.8mL | |
| 1.07m2 | 1.18m2 | 0.9mL | |
| 1.19m2 | 1.31m2 | 1.0mL | |
| 1.32m2 | 1.43m2 | 1.1mL | |
| 1.44m2 | 1.56m2 | 1.2mL | |
| 1.57m2 | 1.68m2 | 1.3mL | |
| 1.69m2 | 1.81m2 | 1.4mL | |
| 1.82m2 | 1.93m2 | 1.5mL | |