医薬品情報
| 総称名 | アトモキセチン |
|---|---|
| 一般名 | アトモキセチン塩酸塩 |
| 欧文一般名 | Atomoxetine Hydrochloride |
| 製剤名 | アトモキセチン塩酸塩内用液 |
| 薬効分類名 | 注意欠陥/多動性障害治療剤(選択的ノルアドレナリン再取り込み阻害剤) |
| 薬効分類番号 | 1179 |
| ATCコード | N06BA09 |
| KEGG DRUG |
D02574
アトモキセチン塩酸塩
|
| JAPIC | 添付文書(PDF) |
添付文書情報2023年12月 改訂(第1版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| アトモキセチン内用液0.4%「トーワ」 (後発品) | ATOMOXETINE ORAL SOLUTION 0.4%"TOWA" | 東和薬品 | 1179050S1030 | 37.2円/mL | 劇薬, 処方箋医薬品注) |
2. 禁忌
次の患者には投与しないこと
4. 効能または効果
注意欠陥/多動性障害(AD/HD)
5. 効能または効果に関連する注意
5.2 AD/HDの診断は、米国精神医学会の精神疾患の診断・統計マニュアル(DSM注))等の標準的で確立した診断基準に基づき慎重に実施し、基準を満たす場合にのみ投与すること。
注)Diagnostic and Statistical Manual of Mental Disorders
6. 用法及び用量
<18歳未満の患者>
通常、18歳未満の患者には、アトモキセチンとして1日0.5mg/kg(0.125mL/kg)より開始し、その後1日0.8mg/kg(0.2mL/kg)とし、さらに1日1.2mg/kg(0.3mL/kg)まで増量した後、1日1.2〜1.8mg/kg(0.3〜0.45mL/kg)で維持する。
ただし、増量は1週間以上の間隔をあけて行うこととし、いずれの投与量においても1日2回に分けて経口投与する。
なお、症状により適宜増減するが、1日量は1.8mg/kg(0.45mL/kg)又は120mg(30mL)のいずれか少ない量を超えないこと。
ただし、増量は1週間以上の間隔をあけて行うこととし、いずれの投与量においても1日2回に分けて経口投与する。
なお、症状により適宜増減するが、1日量は1.8mg/kg(0.45mL/kg)又は120mg(30mL)のいずれか少ない量を超えないこと。
<18歳以上の患者>
通常、18歳以上の患者には、アトモキセチンとして1日40mg(10mL)より開始し、その後1日80mg(20mL)まで増量した後、1日80〜120mg(20〜30mL)で維持する。
ただし、1日80mg(20mL)までの増量は1週間以上、その後の増量は2週間以上の間隔をあけて行うこととし、いずれの投与量においても1日1回又は1日2回に分けて経口投与する。
なお、症状により適宜増減するが、1日量は120mg(30mL)を超えないこと。
ただし、1日80mg(20mL)までの増量は1週間以上、その後の増量は2週間以上の間隔をあけて行うこととし、いずれの投与量においても1日1回又は1日2回に分けて経口投与する。
なお、症状により適宜増減するが、1日量は120mg(30mL)を超えないこと。
7. 用法及び用量に関連する注意
8. 重要な基本的注意
8.1 本剤を投与する医師又は医療従事者は、投与前に患者(小児の場合には患者及び保護者又はそれに代わる適切な者)に対して、本剤の治療上の位置づけ及び本剤投与による副作用発現等のリスクについて、十分な情報を提供するとともに、適切な使用方法について指導すること。
8.2 本剤を長期間投与する場合には、必要に応じて休薬期間を設定するなどして、定期的に有用性の再評価を実施すること。
8.3 臨床試験で本剤投与中の小児患者において、自殺念慮や関連行動が認められているため、本剤投与中の患者ではこれらの症状の発現について注意深く観察すること。[15.1.1参照]
8.4 攻撃性、敵意はAD/HDにおいてしばしば観察されるが、本剤の投与中にも攻撃性、敵意の発現や悪化が報告されている。投与中は、攻撃的行動、敵意の発現又は悪化について観察すること。[15.1.2参照]
8.5 通常量の本剤を服用していた精神病性障害や躁病の既往歴がない患者において、幻覚等の精神病性又は躁病の症状が報告されている。このような症状の発現を認めたら、本剤との関連の可能性を考慮すること。投与中止が適切な場合もある。
8.6 眠気、めまい等が起こることがあるので、本剤投与中の患者には自動車の運転等危険を伴う機械の操作に従事させないよう注意すること。
8.8 本剤は血圧又は心拍数に影響を与えることがあるので、本剤を心血管障害のある患者に投与する際は、循環器を専門とする医師に相談するなど、慎重に投与の可否を検討すること。また、患者の心疾患に関する病歴、突然死や重篤な心疾患に関する家族歴等から、心臓に重篤ではないが異常が認められる、若しくはその可能性が示唆される患者に対して本剤の投与を検討する場合には、投与開始前に心電図検査等により心血管系の状態を評価すること。[2.3、9.1.2-9.1.5、15.1.3参照]
8.9 小児において本剤の投与初期に体重増加の抑制、成長遅延が報告されている。本剤の投与中は小児患者の成長に注意し、身長や体重の増加が思わしくないときは減量又は投与の中断等を考慮すること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 痙攣発作又はその既往歴のある患者
痙攣を起こすことがある。
9.1.2 心疾患(QT延長を含む)又はその既往歴のある患者
9.1.3 先天性QT延長症候群の患者又はQT延長の家族歴のある患者
9.1.4 高血圧又はその既往歴のある患者
9.1.5 脳血管障害又はその既往歴のある患者
9.1.6 起立性低血圧の既往歴のある患者
本剤の投与による起立性低血圧の報告がある。
9.1.7 精神系疾患(精神病性障害、双極性障害)のある患者
行動障害、思考障害又は躁病エピソードの症状が悪化するおそれがある。
9.1.8 排尿困難のある患者
症状を悪化させるおそれがある。
9.2 腎機能障害患者
血中濃度が上昇するおそれがある。[16.6.1参照]
9.3 肝機能障害患者
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。動物実験(ラット)において胎盤通過性が認められている。
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。動物実験(ラット)において乳汁中への移行が認められている。
9.7 小児等
9.8 高齢者
一般に生理機能が低下している。
10. 相互作用
相互作用序文
本剤は、主に肝薬物代謝酵素CYP2D6で代謝される。[16.4.1参照]
薬物代謝酵素用語
CYP2D6
10.1 併用禁忌
| MAO阻害剤 セレギリン塩酸塩(エフピー) ラサギリンメシル酸塩(アジレクト) サフィナミドメシル酸塩(エクフィナ) [2.2参照] | 両薬剤の作用が増強されることがある。MAO阻害剤の投与中止後に本剤を投与する場合には、2週間以上の間隔をあけること。また、本剤の投与中止後にMAO阻害剤を投与する場合は、2週間以上の間隔をあけること。 | 脳内モノアミン濃度が高まる可能性がある。 |
10.2 併用注意
| サルブタモール硫酸塩(静脈内投与等の全身性投与。吸入投与を除く) [16.7.3、16.7.4参照] | 心拍数、血圧が上昇したとの報告があるので、注意して投与すること。 | 心血管系への作用を増強する可能性がある。 |
| β-受容体刺激剤(サルブタモール硫酸塩を除く) | これらの薬剤の心拍数、血圧上昇作用が増強するおそれがあるので、注意して投与すること。 | これらの薬剤の心血管系への作用を増強する可能性がある。 |
| CYP2D6阻害剤 パロキセチン塩酸塩水和物等 [7.1、16.7.5参照] | 本剤の血中濃度が上昇することがあるので、経過を観察しながら時間をかけて本剤を増量すること。 | これらの薬剤のCYP2D6阻害作用により本剤の血中濃度が上昇するおそれがある。 |
| 昇圧作用を有する薬剤 ドパミン塩酸塩等 | これらの薬剤の血圧上昇作用が増強するおそれがあるので、注意して投与すること。 | これらの薬剤の血圧への作用に影響する可能性がある。 |
| ノルアドレナリンに影響する薬剤 三環系抗うつ剤(イミプラミン塩酸塩等) 選択的セロトニン・ノルアドレナリン再取り込み阻害剤 メチルフェニデート塩酸塩等 | これらの薬剤の作用が増強するおそれがあるので、注意して投与すること。 | これらの薬剤のノルアドレナリンへの作用を相加的又は相乗的に増強する可能性がある。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 肝機能障害、黄疸、肝不全(いずれも頻度不明)
肝機能検査値の上昇を伴う肝機能障害、黄疸、肝不全があらわれることがある。
11.1.2 アナフィラキシー(頻度不明)
血管神経性浮腫、蕁麻疹等のアナフィラキシーがあらわれることがある。
11.2 その他の副作用
| 5%以上 | 1〜5%未満 | 1%未満 | 頻度不明 | |
| 消化器 | 悪心(31.5%)、食欲減退(19.9%)、腹痛、嘔吐、便秘、口渇 | 下痢、消化不良、口内乾燥 | 鼓腸 | |
| 精神神経系 | 頭痛(15.4%)、傾眠(15.8%)、浮動性めまい、不眠症 | 体位性めまい、睡眠障害、易刺激性、不快気分 | 早朝覚醒型不眠症、気分変化、振戦、抑うつ気分、錯感覚、不安、感覚鈍麻、幻覚を含む感覚障害、うつ病、攻撃性、リビドー減退、チック、激越、落ち着きのなさ | びくびく感 |
| 過敏症 | そう痒症 | 発疹、蕁麻疹 | ||
| 循環器 | 動悸 | 頻脈、血圧上昇、心拍数増加 | 心電図QT延長、失神 | レイノー現象、潮紅 |
| 皮膚 | 多汗症 | 皮膚炎 | ||
| 泌尿・生殖器 | 排尿困難、勃起不全、不規則月経 | 生殖器痛、尿閉、月経困難症、射精障害、前立腺炎、頻尿 | 持続勃起、勃起時疼痛、射精不能、精巣痛、オルガズム異常、尿意切迫 | |
| その他 | 体重減少 | 胸痛、無力症、疲労、ほてり、悪寒、味覚異常 | 結膜炎、胸部不快感、末梢冷感、冷感、筋痙縮 | 散瞳 |
13. 過量投与
13.1 症状
過量投与時には、痙攣、QT延長、傾眠、興奮、運動亢進、異常行動、消化器症状、散瞳、頻脈、口渇、浮動性めまい、振戦及び血圧上昇等が認められている。また、本剤及び他剤を同時に過量投与した場合には、死亡例も報告されている。
13.2 処置
本剤は蛋白結合率が高いため、透析は有効ではない。
14. 適用上の注意
14.1 薬剤投与時の注意
内服用にのみ使用させること。
14.2 薬剤交付時の注意
14.2.1 本剤を希釈しないこと。本剤は瓶包装品のまま交付すること。やむを得ず本剤を小分けする場合は、本剤の専用容器を使用すること。また、患者(小児の場合には患者及び保護者又はそれに代わる適切な者)に対し、本剤に添付されている使用説明書を渡し、服用方法を指導すること。
14.2.2 小児の手の届かない所に保管するよう指導すること。
14.2.3 眼球刺激性があるため、内用液が眼球に付着した場合はすぐに水で洗浄し、医師に相談するよう指導すること。また、手やその他の付着した可能性のある箇所は、すぐ水で洗浄するよう指導すること。
15. その他の注意
15.1 臨床使用に基づく情報
15.1.1 外国の小児及び青少年を対象としたプラセボ対照短期試験(AD/HD患者における11試験及び遺尿症患者における1試験の計12試験)の併合解析において、プラセボ投与群に対してアトモキセチン投与群では投与初期の自殺念慮のリスクが大きかったとの報告がある(アトモキセチン投与群5/1357(0.37%)、プラセボ投与群0/851(0%))。なお、これらの試験において既遂例は認められなかった。また、AD/HDに併存する精神系疾患は自殺念慮、自殺行動のリスクの増加に関連しているとの外国の報告がある。[8.3参照]
15.1.2 外国の小児及び青少年を対象としたプラセボ対照短期試験(AD/HD患者における11試験)の併合解析において、攻撃的行動、敵意の発現率はアトモキセチン投与群21/1308(1.6%)、プラセボ投与群9/806(1.1%)であった。日本及び外国の成人を対象としたプラセボ対照短期試験(AD/HD患者における9試験)の併合解析において、攻撃的行動、敵意の発現率はアトモキセチン投与群6/1697(0.35%)、プラセボ投与群4/1560(0.26%)であった。[8.4参照]
15.2 非臨床試験に基づく情報
15.2.1 幼若ラットにアトモキセチン1、10及び50mg/kgを約75日間反復投与したところ、1mg/kg以上で性成熟のわずかな遅延、10mg/kg以上で精巣上体尾部重量の低下及び精巣上体中の精子数減少が見られたが、性成熟後の生殖能や受胎能に影響はなかった。ラットで生じたこれらの変化は軽度であったが、そのときの血漿中濃度(AUC)を臨床最大用量投与時(1.8mg/kg)のAUCと比較すると1mg/kgでは最大で0.2倍(CYP2D6通常活性、EM)又は0.02倍(CYP2D6活性欠損、PM)、10mg/kgでは最大で1.9倍(EM)又は0.2倍(PM)であり、臨床用量での安全域は確保されていない。なお、外国の小児及び青少年患者において、第二次性徴に対する影響を調べた臨床試験ではアトモキセチン投与の性成熟に対する影響は示唆されなかった。
15.2.2 妊娠ウサギに器官形成期を通じてアトモキセチンを経口投与した3試験のうち1試験において、最高用量の100mg/kgで生存胎児数の減少、早期吸収胚の増加、総頚動脈起始異常と鎖骨下動脈欠損の発現率の微増が認められたが、これらの変化は背景データの範囲内であった。この用量では軽度の体重増加の抑制及び摂餌量の低下等の母体毒性も認められており、このときのAUCは臨床最大用量投与時(1.8mg/kg)のAUCと比較すると2.6倍(EM)又は0.3倍(PM)であった。なお、これらの所見が認められたのは3試験のうち1試験であり、アトモキセチン投与との関連性及びヒトへの外挿性は不明である。
16. 薬物動態
16.1 血中濃度
16.1.1 CYP2D6の遺伝子型の解析
本臨床評価に際し、CYP2D6活性を遺伝子型により分類し、不活性型アレルをホモで有する場合を不活性(Poor Metabolizer、PM)、それ以外を通常活性(Extensive Metabolizer、EM)と定義した。日本人ではPMの割合が少ないことから、EMを更に細分化し、CYP2D6の活性が低下した遺伝子が関連するIntermediate Metabolizer(IM)を定義した。1)
| CYP2D6 表現型 | CYP2D6 表現型の詳細分類 | CYP2D6遺伝子型注1) (アレル/アレル) |
| PM | PM | 不活性型/不活性型 |
| EM | UM(Ultra rapid Metabolizer) | 通常活性型/通常活性型注2) |
| EM | 通常活性型/通常活性型 | |
| IM | 通常活性型/活性低下型 通常活性型/不活性型 活性低下型/活性低下型 活性低下型/不活性型 |
16.1.2 単回投与
CYP2D6 EM健康成人にアトモキセチン10、40、90又は120mgを単回経口投与注17)したときの最高血漿中濃度(Cmax)及び血漿中濃度曲線下面積(AUC)は、投与量に比例して増加した。2)
| 投与量 | AUC0-∞ (μg・hr/mL) | Cmax (ng/mL) | Tmax (hr)注3) | T1/2 (hr)注4) | CL/F (L/hr) |
| 10mg (n=22) | 0.574(70.2) | 110.53(33.2) | 1.25 (0.50〜2.00) | 3.46 (1.85〜6.61) | 22.93(43.0) |
| 40mg (n=21) | 2.51(68.5) | 478.36(33.5) | 1.00 (0.50〜4.00) | 4.12 (2.09〜7.06) | 21.18(47.0) |
| 90mg (n=20) | 5.30(54.2) | 920.03(33.1) | 1.75 (0.50〜6.00) | 4.01 (2.16〜7.03) | 20.50(39.3) |
| 120mg (n=19) | 6.43(37.5) | 1086.23(30.6) | 1.00 (0.50〜4.00) | 4.27 (2.86〜6.23) | 21.43(38.7) |
16.1.3 反復投与
CYP2D6 EM健康成人にアトモキセチン1回40mg又は60mgを1日2回7日間反復経口投与注17)したときの血漿中濃度は、初回投与約1時間後にそれぞれCmax427.34ng/mL及び615.52ng/mLに達した。反復投与開始から約24時間で定常状態に達すると予測され、反復投与時において最終投与約1時間後にCmax604.52ng/mL及び874.33ng/mLに達した。2)
| 投与量 | 投与 | AUC0-12 (μg・hr/mL) | Cmax(ng/mL) | Tmax(hr)注5) |
| 40mg(n=10) | 初回 | 1.95(38.3) | 427.34(33.9) | 1.25 (0.50〜2.00) |
| 40mg(n=10) | 反復 | 2.47(42.0)注6) | 604.52(35.3) | 1.00 (0.50〜1.50) |
| 60mg(n=10) | 初回 | 3.14(41.6) | 615.52(32.3) | 1.00 (1.00〜2.00) |
| 60mg(n=9) | 反復 | 3.73(41.8)注6) | 874.33(26.2) | 1.00 (0.50〜2.00) |
16.1.4 成人と患児の薬物動態比較
CYP2D6 EM健康成人と患児(7〜14歳)の薬物動態を比較した結果を示した。患児と成人のCmax(投与量を体重で補正)及び消失半減期は同程度であることが示された。体重補正したクリアランスと分布容積にも両者間で大きな違いは認められなかった(外国人データ)。3)
| 集団 | Cmax注7) (ng/mL)/(mg/kg) | Cmax,ss注7) (ng/mL)/(mg/kg) | T1/2(hr) | CL/F (L/hr/kg) | Vz/F (L/kg) |
| 患児EM | 512 | 524 | 3.19 | 0.435 | 2.01 |
| 成人EM | 569 | 667 | 3.56 | 0.352 | 1.82 |
16.1.5 生物学的同等性試験
アトモキセチン内用液0.4%「トーワ」とストラテラ内用液0.4%を、クロスオーバー法によりそれぞれ10mL(アトモキセチンとして40mg)クリアランスの大きな健康成人男子に絶食単回経口投与して血漿中未変化体濃度を測定し、得られた薬物動態パラメータ(AUC、Cmax)について90%信頼区間法にて統計解析を行った結果、log(0.80)〜log(1.25)の範囲内であり、両剤の生物学的同等性が確認された。4)
| 判定パラメータ | 参考パラメータ | |||
| AUC0-24 (ng・hr/mL) | Cmax (ng/mL) | Tmax (hr) | T1/2 (hr) | |
| アトモキセチン内用液 0.4%「トーワ」 | 2086±859 | 383.2±141.8 | 1.44±1.67 | 3.32±0.80 |
| ストラテラ内用液0.4% | 2000±834 | 376.4±132.2 | 0.75±0.29 | 3.33±0.81 |
血漿中濃度並びにAUC、Cmax等のパラメータは、被験者の選択、体液の採取回数・時間等の試験条件によって異なる可能性がある。
16.2 吸収
16.2.1 絶対的生物学的利用率
CYP2D6 EM及びPM健康成人における絶対的生物学的利用率はそれぞれ約63%及び94%であった(外国人データ)。5)
16.2.2 食事の影響
CYP2D6 EM健康成人にアトモキセチン40mg又は60mgを空腹時又は食後に単回経口投与注17)したとき、高脂肪食摂取によって空腹時に比較してCmaxは37%減少し、Tmaxは約2時間遅延したが、AUCには差は認められなかった。CYP2D6 EM患児における母集団薬物動態解析の結果では、食事によるCmaxの減少は9%であった(外国人データ)。6)
16.3 分布
アトモキセチン静脈内投与後の分布容積は0.85L/kg(CYP2D6 EM健康成人)及び0.91L/kg(CYP2D6 PM健康成人)であり、主に全体液中に広く分布すると考えられた(外国人データ)。
アトモキセチン濃度150〜3000ng/mLの範囲において、in vitroヒト血漿蛋白結合率は約98%であり、主にアルブミンに結合する。7)
16.4 代謝
16.4.1 代謝酵素及び代謝物
アトモキセチンは主に薬物代謝酵素CYP2D6によって代謝される。主要酸化代謝物は4-ヒドロキシ体であり、これはすぐにグルクロン酸抱合化される。4-ヒドロキシ体はアトモキセチンとほぼ同等のノルアドレナリン取り込み阻害作用を有するが血漿中濃度は非常に低い。4-ヒドロキシ体は主にCYP2D6により生成されるが、CYP2D6活性が欠損していても、他の数種のCYP酵素から低速ながら生成される(外国人データ)。また、CYP2D6活性が欠損した被験者から得たヒト肝ミクロソームを用いたin vitro試験では、アトモキセチンとCYP2D6阻害剤を併用しても4-ヒドロキシ体生成に対して阻害は認められなかった。ヒト肝ミクロソーム及び培養肝細胞を用いたin vitro試験により、アトモキセチンはCYP1A2又はCYP3Aを誘導しないこと、CYP1A2、CYP3A、CYP2D6又はCYP2C9を阻害しないことが確認された。8)[7.1、10.参照]
16.4.2 CYP2D6遺伝子多型の薬物動態に及ぼす影響
外国のPM健康成人では、EM健康成人に比較して定常状態のアトモキセチンの平均血漿中濃度(Cav,ss)が約10倍、定常状態のCmax,ssが約5倍高値であった。9)
| 遺伝子型 | Cav,ss (ng/mL)/ (mg/kg)注8) | Cmax,ss (ng/mL)/ (mg/kg)注8) | Tmax(hr)注9) | T1/2(hr) | CL/F(L/hr/kg) |
| EM(n=223) | 249(58.5) | 667(41.3) | 1.00(0.50,2.00) | 3.56(27.5) | 0.352(55.7) |
| PM(n=28) | 2540(14.0) | 3220(11.3) | 2.50(1.00,6.00) | 20.6(17.3) | 0.0337(18.8) |
日本人において、EMを更に3つに分類した場合(UM、EM及びIM注10))、IM注10)のAUCの算術平均値はEM注10)に比較して約1.4倍高値であった。なお、日本人にはUMは該当がなかった。10)[7.1、9.1.9参照]
| 遺伝子型 | AUC0-∞ (μg・hr/mL) | Cmax (ng/mL) | T1/2(hr)注11) |
| EM注10)(n=5) | 4.95(39.4) | 861(23.3) | 3.87(2.85〜4.87) |
| IM注10)(n=14) | 6.96(34.4) | 1170(28.9) | 4.41(3.04〜6.23) |
16.5 排泄
健康成人統合解析におけるアトモキセチンの平均消失半減期は、CYP2D6 EM及びPMでそれぞれ3.6時間及び20.6時間であった。
健康成人にアトモキセチン1回20mgを1日2回5日反復経口投与注15)した後に、14C標識アトモキセチン20mgを単回経口投与したときの放射能は、CYP2D6 EMでは投与後168時間以内に投与量の約96%が尿中にほとんど代謝物として排泄され、糞中には約2%が排泄された。CYP2D6 PMでは、投与後264時間以内に投与した放射能の約80%が尿中にほとんど代謝物として排泄され、糞中には約17%が排泄された。また、尿中から回収された放射能のうち、未変化体は約1%(EM)及び約2%(PM)であり、主代謝物の4-ヒドロキシアトモキセチン-O-グルクロン酸抱合体は84%(EM)及び31%(PM)であった(外国人データ)。11)
| 尿 | 糞 | 尿糞 | |
| EM(n=4)注12) | 95.81±2.16 | 1.67±0.32 | 97.48±1.92 |
| PM(n=3)注13) | 79.92±2.39 | 16.91±2.50 | 96.83±1.09 |
16.6 特定の背景を有する患者
16.6.1 腎機能障害時の血漿中濃度
CYP2D6 EMの成人腎不全患者にアトモキセチン20mgを単回経口投与注17)したとき、末期腎不全患者において、健康成人に比較して64%のAUCの増大が認められたが、体重で補正した投与量に換算することによって、その差は24%になった(外国人データ)。12)[9.2参照]
| AUC0-∞ (μg・hr/mL) | AUC0-∞ (μg・hr/mL)/(mg/kg)注14) | Cmax (ng/mL) | Cmax (ng/mL)/(mg/kg)注14) | |
| 健康成人(n=6) | 0.469 | 2.26 | 86.0 | 415 |
| 腎不全患者(n=6) | 0.769 | 2.80 | 92.2 | 336 |
16.6.2 肝機能障害時の血漿中濃度
CYP2D6 EMの成人肝硬変患者にアトモキセチン20mgを単回経口投与注17)したとき、中等度(Child-Pugh分類B)及び重度(Child-Pugh分類C)肝硬変患者において、それぞれ健康成人に比較してAUCが約2倍及び約4倍に増大した(外国人データ)。13)[7.2、9.3参照]
| AUC0-∞ (μg・hr/mL) | Cmax (ng/mL) | Tmax (hr)注15) | T1/2 (hr)注16) | CL/F (L/hr/kg) | |
| 健康成人 (n=10) | 0.706 (67.9) | 142 (36.0) | 1.02 (0.50〜1.55) | 4.26 (2.35〜8.03) | 0.506 (53.5) |
| 中等度肝硬変患者 (n=6) | 1.17 (36.7) | 116 (55.2) | 3.27 (0.50〜6.00) | 11.0 (7.85〜17.9) | 0.208 (28.1) |
| 重度肝硬変患者 (n=4) | 2.73 (63.0) | 126 (44.8) | 5.98 (0.50〜12.02) | 16.0 (7.21〜26.3) | 0.155 (78.5) |
16.7 薬物相互作用
16.7.1 蛋白結合率の高い薬剤との併用
アトモキセチンは、治療濃度のアセチルサリチル酸、ジアゼパム、フェニトイン、ワルファリンのヒト血漿蛋白結合率に影響を及ぼさなかった。同様に上記薬剤は、アトモキセチンのヒト血漿蛋白結合率に影響を及ぼさなかった(in vitro)。14)
16.7.2 メチルフェニデートとの併用
CYP2D6 EM健康成人にメチルフェニデート60mgを1日1回5日間経口投与し、アトモキセチン60mgを3、4、5日目に1日2回3日間経口投与注17)したとき、アトモキセチンとメチルフェニデートの併用により、メチルフェニデート単剤投与時に認められた心拍数及び収縮期・拡張期血圧への影響は増強しなかった(外国人データ)。15)
16.7.3 吸入サルブタモールとの併用
16.7.4 サルブタモール静脈内投与との併用
16.7.5 CYP2D6阻害剤との併用
CYP2D6 EMの健康成人にパロキセチン20mgを1日1回経口投与時の定常状態で、アトモキセチン20mgを1日2回反復経口投与注17)したとき、パロキセチンとの併用により、定常状態におけるアトモキセチンのCmax及びAUCはそれぞれ約3.5倍及び約6.5倍に増加し、そのときの血中濃度はCYP2D6 PM健康成人にアトモキセチンを単剤投与したときの血中濃度と同程度であった(外国人データ)。18)
| AUC0-12 (μg・hr/mL) | Cmax(ng/mL) | T1/2(hr) | |
| アトモキセチン単剤(n=21) | 0.77 | 173 | 3.92 |
| パロキセチン併用(n=14) | 5.01 | 612 | 10.0 |
16.7.6 胃のpHに影響する薬剤との併用
CYP2D6 EM健康成人にアトモキセチン40mg単回経口投与注17)、あるいはオメプラゾール80mg又はマグネシウム/アルミニウム水酸化物20mLを併用投与したとき、アトモキセチンの生物学的利用率は変化しなかった(外国人データ)。20)
16.7.7 ミダゾラムとの併用
CYP2D6 PM健康成人にアトモキセチン60mgを1日2回12日間経口投与注17)し、CYP3A4の基質であるミダゾラム5mgを単回経口投与したとき、ミダゾラムのCmaxとAUC0-∞は約16%増加したが被験者内変動に含まれるものであった(外国人データ)。21)
16.7.8 エタノールとの併用
CYP2D6 EM健康成人及びCYP2D6 PM健康成人にアトモキセチン40mgを1日2回5日間経口投与注17)し、エタノール2.0mL/kg(0.6mg/kg)を単回経口投与したとき、疲労スケール、複合鎮静スコア、継続的注意力で示されるエタノールの中枢作用をアトモキセチンは増強も減弱もしなかった(外国人データ)。22)
注17)本剤の承認された用法・用量は、「小児:1日0.5mg/kg(0.125mL/kg)より開始し、その後1日0.8mg/kg(0.2mL/kg)とし、さらに1日1.2mg/kg(0.3mL/kg)まで増量した後、1日1.2〜1.8mg/kg(0.3〜0.45mL/kg)で維持する。成人:1日40mg(10mL)より開始し、その後1日80mg(20mL)まで増量した後、1日80〜120mg(20〜30mL)で維持する。」である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
<小児AD/HD患者>
17.1.1 国内第II/III相試験
小児AD/HD患者(6歳以上18歳未満)を対象に実施したプラセボ対照二重盲検群間比較試験において、有効性の評価尺度であるADHD RS-IV日本語版(医師用)総スコアは下表のとおりであった。
| 投与群 | N | ベースライン | 最終観察時 | 変化量 | 差注1) | 95%信頼区間注1) | p値注2) Williams | ||||
| 平均 | 標準偏差 | 平均 | 標準偏差 | 平均 | 標準偏差 | 信頼下限 | 信頼上限 | ||||
| プラセボ | 61 | 32.3 | 9.6 | 24.2 | 11.4 | -8.1 | 7.1 | ||||
| ATX 0.5注6) | 62 | 32.3 | 8.4 | 22.7 | 11.4 | -9.6 | 9.1 | -1.5 | -4.3 | 1.3 | − |
| ATX 1.2 | 58 | 33.3 | 8.7 | 22.5 | 10.3 | -10.8 | 6.8 | -2.5 | -5.4 | 0.3 | 0.037 |
| ATX 1.8 | 60 | 31.5 | 7.8 | 19.8 | 9.0 | -11.6 | 8.8 | -3.7 | -6.5 | -0.8 | 0.010 |
17.1.2 国内第III相長期継続投与試験
小児AD/HD患者(6歳以上18歳未満)を対象に実施したプラセボ対照二重盲検群間比較試験を完了した小児患者を対象に実施した長期継続投与試験において、有効性の評価尺度であるADHD RS-IV日本語版(医師用)総スコアの推移は下表のとおりであった。
| 期間(月) | N | 平均 | 標準偏差 |
| 0 | 228 | 22.2 | 10.4 |
| 0.5 | 221 | 21.7 | 10.2 |
| 1 | 204 | 19.7 | 9.8 |
| 3 | 206 | 16.4 | 9.6 |
| 6 | 169 | 14.8 | 9.6 |
| 12 | 146 | 12.7 | 8.4 |
17.1.3 外国第III相試験
外国の小児AD/HD患者(8歳以上18歳未満)を対象に実施したプラセボ対照二重盲検群間比較試験において、有効性の評価尺度であるADHD RS-IV-Parent:Inv総スコアは下表のとおりであった。
| 投与群 | N | ベースライン | 最終観察時 | 変化量 | p値注3) | |||
| 平均 | 標準偏差 | 平均 | 標準偏差 | 平均 | 標準偏差 | |||
| プラセボ | 83 | 38.3 | 8.9 | 32.5 | 13.8 | -5.8 | 10.9 | |
| ATX 0.5注6) | 43 | 40.2 | 9.6 | 30.3 | 15.2 | -9.9 | 14.6 | |
| ATX 1.2 | 84 | 39.2 | 9.2 | 25.5 | 13.8 | -13.6 | 14.0 | <0.001 |
| ATX 1.8 | 82 | 39.7 | 8.7 | 26.2 | 14.8 | -13.5 | 14.5 | <0.001 |
<成人AD/HD患者>
17.1.4 国際共同第III相試験
成人AD/HD患者(18歳以上)を対象に実施したプラセボ対照二重盲検群間比較試験において、有効性の評価尺度であるCAARS-inv:SV AD/HD症状総スコアは下表のとおりであった。
| 投与群 | N | ベースライン | 最終観察時 | 変化量 | 差注4) | 95%信頼区間注4) | p値注4) | ||||
| 平均 | 標準偏差 | 平均 | 標準偏差 | 平均 | 標準偏差 | 信頼下限 | 信頼上限 | ||||
| プラセボ | 195 | 33.9 | 7.5 | 25.1 | 11.2 | -8.8 | 9.6 | ||||
| ATX | 191 | 33.2 | 7.8 | 18.9 | 10.2 | -14.3 | 10.4 | -5.78 | -7.66 | -3.91 | <0.001 |
アトモキセチンを投与された193例中142例(73.6%)に副作用が認められた。主な副作用(10%以上)は悪心(40.4%、78/193例)、食欲減退(22.8%、44/193例)、傾眠(15.0%、29/193例)、口内乾燥(10.4%、20/193例)であった。28)
17.1.5 国際共同第III相長期継続投与試験
成人AD/HD患者(18歳以上)を対象に実施したプラセボ対照二重盲検群間比較試験を完了した患者を対象に実施した長期継続投与試験において、有効性の評価尺度であるCAARS-inv:SV AD/HD症状総スコアの推移は下表のとおりであった。
| 期間(月) | N | 平均 | 標準偏差 |
| 0 | 211 | 22.2 | 10.9 |
| 0.5 | 211 | 20.9 | 10.6 |
| 1 | 206 | 19.3 | 9.9 |
| 1.5 | 203 | 17.9 | 9.8 |
| 2 | 200 | 16.7 | 9.5 |
| 3 | 190 | 16.4 | 9.3 |
| 4 | 175 | 15.4 | 9.3 |
| 5 | 173 | 14.8 | 9.1 |
| 6 | 163 | 15.0 | 9.6 |
| 7 | 155 | 14.1 | 9.0 |
| 8 | 149 | 13.5 | 9.4 |
| 9 | 144 | 14.3 | 9.5 |
| 10 | 140 | 13.8 | 9.4 |
| 11 | 138 | 13.2 | 9.3 |
| 12 | 135 | 13.1 | 9.5 |
アトモキセチンを投与された211例中139例(65.9%)に副作用が認められた。主な副作用(10%以上)は悪心(42.2%、89/211例)、口渇(12.8%、27/211例)であった。29)
17.3 その他
17.3.1 QT間隔に対する作用
CYP2D6 PM健康成人(131例)に、アトモキセチン20mg、アトモキセチン60mg、プラセボをそれぞれ1日2回反復経口投与注6)、モキシフロキサシン400mg(陽性対照)単回経口投与の4期クロスオーバーのtQT試験を行った。血中アトモキセチン濃度の上昇に伴いわずかにQTcM間隔(時点を一致させたベースラインからのQT間隔変化量を応答変数、時間を一致させたベースラインからのRR間隔変化量、時間、治療及び時間×治療を固定効果、被験者、被験者×時間及び被験者×治療を変量効果とする混合効果モデルにより算出した)の延長が認められたが、臨床使用で想定される最高血中濃度においてもアトモキセチンのQTc間隔に対する影響はプラセボと比較して臨床的に意義のある差ではなかった(外国人データ)。
| 投与量 | 投与後時間 (hr) | プラセボとの差 [90%信頼区間](msec) |
| アトモキセチン20mgBID | 2 | 0.5[-1.2,2.2] |
| アトモキセチン60mgBID | 2 | 4.2[2.5,6.0] |
| モキシフロキサシン400mg注5) | 4 | 4.8[3.3,6.4] |
注6)本剤の承認された用法・用量は、「小児:1日0.5mg/kg(0.125mL/kg)より開始し、その後1日0.8mg/kg(0.2mL/kg)とし、さらに1日1.2mg/kg(0.3mL/kg)まで増量した後、1日1.2〜1.8mg/kg(0.3〜0.45mL/kg)で維持する。成人:1日40mg(10mL)より開始し、その後1日80mg(20mL)まで増量した後、1日80〜120mg(20〜30mL)で維持する。」である。
18. 薬効薬理
18.1 作用機序
臨床における有用性には神経終末のノルアドレナリントランスポーターに対する選択的阻害作用が関与していることが可能性としては考えられるものの、明確な機序は不明である。31)
18.2 薬理作用
18.2.1 モノアミン取り込み阻害作用(in vitro)
アトモキセチンはラット脳シナプトソームへのノルアドレナリン取り込みを強力に阻害した(Ki:4.47nM)。アトモキセチンのノルアドレナリン取り込み阻害作用はセロトニン及びドパミン取り込み阻害作用に比較して30倍以上選択的であった。なお、アトモキセチンは各種神経伝達物質受容体にはほとんど親和性を示さなかった。32)
18.2.2 モノアミン取り込み阻害作用(in vivo)
アトモキセチンは神経毒によるノルアドレナリン枯渇を阻害したが(ED50:2.5mg/kg,p.o.)、セロトニン枯渇に対してはほとんど作用を示さなかった。33)
18.2.3 細胞外モノアミン濃度に及ぼす影響(in vivo)
アトモキセチンは前頭前野におけるノルアドレナリン及びドパミンの細胞外濃度を有意に上昇させたが(0.3〜3mg/kg,i.p.)、線条体や側坐核における細胞外ドパミン濃度には影響を及ぼさなかった。33)
19. 有効成分に関する理化学的知見
19.1. アトモキセチン塩酸塩
| 一般的名称 | アトモキセチン塩酸塩 |
|---|---|
| 一般的名称(欧名) | Atomoxetine Hydrochloride |
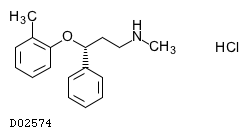
| 化学名 | (3R)-N-Methyl-3-(2-methylphenoxy)-3-phenylpropan-1-amine monohydrochloride |
| 分子式 | C17H21NO・HCl |
| 分子量 | 291.82 |
| 物理化学的性状 | 白色の粉末である。ジメチルスルホキシドに溶けやすく、エタノール(99.5)にやや溶けやすく、水にやや溶けにくい。 |
| KEGG DRUG |
|
22. 包装
100mL×1瓶
23. 主要文献
- CYP2D6の遺伝子型の解析(ストラテラカプセル:2012年8月24日承認、申請資料概要2.7.2.1)
- 日本人健康成人を対象とした単回投与及び反復投与試験(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.6.2)
- 成人と患児の薬物動態比較(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.2.3)
- 麦谷 歩ほか, 診療と新薬, 55 (9), 649-659, (2018)
- 絶対的バイオアベイラビリティ(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.6.2)
- バイオアベイラビリティに対する食事の影響(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.1.3)
- 分布(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.2.3)
- 代謝、薬物相互作用(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.2、2.7.2.3)
- 外国人PM健康成人における薬物動態(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.2.3、2.7.2.5)
- 日本人健康成人における薬物動態(ストラテラカプセル:2012年8月24日、申請資料概要2.7.2.2)
- 排泄(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.2.2、2.7.2.3)
- 末期腎不全患者における薬物動態(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.2.2、2.7.2.5)
- 肝硬変患者における薬物動態(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.6.2)
- 薬物相互作用(in vitro)(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.2)
- 経口メチルフェニデート併用における血行力学パラメータに及ぼす影響(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.6.2)
- 吸入サルブタモール併用における血行力学パラメータの変化(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.6.2)
- サルブタモール静脈内投与併用における血行力学パラメータの変化(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.6.2)
- パロキセチン併用における安全性及び薬物動態学的相互作用(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.6.2)
- フルオキセチン併用における安全性及び薬物動態学的相互作用(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.6.2)
- オメプラゾール及びマグネシウム/アルミニウム水酸化物併用における相対的バイオアベイラビリティ(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.6.2)
- ミダゾラム併用における安全性及び薬物動態学的相互作用(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.6.2)
- エタノール併用における精神運動作用(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.6.2)
- Takahashi M,et al., J.Child Adolesc.Psychopharmacol., 19 (4), 341-351, (2009) »PubMed
- 小児AD/HD患者に対する国内第II/III相試験(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.6.3)
- 小児AD/HD患者に対する第III相長期継続投与臨床試験(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.6.3)
- Michelson D,et al., Pediatrics., 108 (5), e83, (2001) »PubMed
- 小児AD/HD患者に対する外国第III相試験(ストラテラカプセル:2009年4月22日承認、申請資料概要2.7.4.2、2.7.6.3)
- 成人AD/HD患者に対する第III相短期投与プラセボ対照二重盲検比較試験(ストラテラカプセル:2012年8月24日承認、申請資料概要2.5.4.2、2.7.6.3)
- 成人AD/HD患者に対する第III相長期継続投与臨床試験(ストラテラカプセル:2012年8月24日承認、申請資料概要2.5.4.2、2.7.6.4)
- Corina Loghin,et al., Br J Clin Pharmacol., 75 (2), 538-549, (2013)
- 作用機序(ストラテラカプセル:2009年4月22日承認、申請資料概要2.6.1)
- モノアミン取り込み阻害作用(ストラテラカプセル:2009年4月22日承認、申請資料概要2.6.2.2、2.6.2.3)
- Bymaster FP,et al., Neuropsychopharmacology., 27 (5), 699-711, (2002) »PubMed
24. 文献請求先及び問い合わせ先
文献請求先
東和薬品株式会社
学術部DIセンター
〒570-0081
大阪府守口市日吉町2丁目5番15号
電話:0120-108-932
FAX:06-7177-7379
製品情報問い合わせ先
東和薬品株式会社
学術部DIセンター
〒570-0081
大阪府守口市日吉町2丁目5番15号
電話:0120-108-932
FAX:06-7177-7379
26. 製造販売業者等
26.1 製造販売元
東和薬品株式会社
大阪府門真市新橋町2番11号