医薬品情報
| 総称名 | ドウベイト |
|---|---|
| 一般名 | ドルテグラビルナトリウム ラミブジン |
| 欧文一般名 | Dolutegravir Sodium Lamivudine |
| 製剤名 | ドルテグラビルナトリウム・ラミブジン配合錠 |
| 薬効分類名 | 抗ウイルス化学療法剤 |
| 薬効分類番号 | 6250 |
| ATCコード | J05AR25 |
| KEGG DRUG |
D11522
ドルテグラビル・ラミブジン
|
| KEGG DGROUP |
DG03107
抗HIV薬
|
| JAPIC | 添付文書(PDF) |
添付文書情報2024年8月 改訂(第8版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ドウベイト配合錠 | Dovato Combination Tablets | ヴィーブヘルスケア | 6250119F1029 | 4792円/錠 | 劇薬, 処方箋医薬品 |
1. 警告
1.1 膵炎を発症する可能性のある小児の患者(膵炎の既往歴のある小児、膵炎を発症させることが知られている薬剤との併用療法を受けている小児)では、本剤の適用を考える場合には、他に十分な効果の認められる治療法がない場合にのみ十分注意して行うこと。これらの患者で膵炎を疑わせる重度の腹痛、悪心・嘔吐等又は血清アミラーゼ、血清リパーゼ、トリグリセリド等の上昇があらわれた場合は、本剤の投与を直ちに中止すること。[8.4、9.1.1、9.7.2、11.1.3参照]
1.2 B型慢性肝炎を合併している患者では、ラミブジンの投与中止により、B型慢性肝炎が再燃するおそれがあるので、本剤の投与を中断する場合には十分注意すること。特に非代償性の場合、重症化するおそれがあるので注意すること。[9.1.2参照]
2. 禁忌
次の患者には投与しないこと
本剤の成分に対し過敏症の既往歴のある患者
4. 効能または効果
5. 効能または効果に関連する注意
5.1 以下のいずれかのヒト免疫不全ウイルス(HIV)感染症患者に使用すること。
・ウイルス学的失敗の経験がなく、切り替え前6ヵ月間以上においてウイルス学的抑制(HIV-1 RNA量が50copies/mL未満)が得られており、本剤の有効成分に対する耐性関連変異を持たず、本剤への切り替えが適切であると判断される抗HIV薬既治療患者。[17.1.3参照]
5.2 本剤による治療にあたっては、可能な場合には薬剤耐性検査(遺伝子型解析あるいは表現型解析)を参考にすること。
6. 用法及び用量
通常、成人及び12歳以上かつ体重40kg以上の小児には、1回1錠(ドルテグラビルとして50mg及びラミブジンとして300mg)を食事の有無に関わらず1日1回経口投与する。
7. 用法及び用量に関連する注意
7.2 本剤とカルバマゼピン、リファンピシン、フェニトイン、ホスフェニトイン、フェノバルビタール、セイヨウオトギリソウ(セント・ジョーンズ・ワート)含有食品を併用する場合は、ドルテグラビルとして50mgを本剤投与の約12時間後に投与する。[7.1、10.2、16.7.1参照]
7.3 本剤はラミブジンの固定用量を含有する配合剤であるので、本剤に加えてラミブジン製剤を併用投与しないこと。
8. 重要な基本的注意
8.1 本剤による治療は、抗HIV療法に十分な経験を持つ医師のもとで開始すること。
8.2 本剤の使用に際しては、国内外のガイドライン等の最新の情報を参考に、患者又は患者に代わる適切な者に、次の事項についてよく説明し同意を得た後、使用すること。
・本剤はHIV感染症の根治療法薬ではないことから、日和見感染を含むHIV感染症の進展に伴う疾病を発症し続ける可能性があるので、本剤投与開始後の身体状況の変化については、すべて担当医に報告すること。
・本剤は併用薬と相互作用を起こすことがあるため、服用中のすべての薬剤等(サプリメントを含む)を担当医に報告すること。また、本剤で治療中に新たに他の薬剤等を服用する場合には、事前に担当医に報告すること。
・担当医の指示なしに用量を変更したり、服用を中止したりしないこと。
・本剤の長期投与による影響については、現在のところ不明であること。
8.3 抗HIV薬の多剤併用療法を行った患者で、免疫再構築炎症反応症候群が報告されている。投与開始後、免疫機能が回復し、症候性のみならず無症候性日和見感染症(マイコバクテリウムアビウムコンプレックス、サイトメガロウイルス、ニューモシスチス等によるもの)等に対する炎症反応が発現することがある。また、免疫機能の回復に伴い自己免疫疾患(甲状腺機能亢進症、多発性筋炎、ギラン・バレー症候群、ブドウ膜炎等)が発現するとの報告があるので、これらの症状を評価し、必要時には適切な治療を考慮すること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 膵炎を発症する可能性のある患者(膵炎の既往歴のある患者、膵炎を発症させることが知られている薬剤との併用療法を受けている患者)
9.1.2 B型慢性肝炎を合併している患者
本剤の投与を中断する場合には十分注意すること。B型慢性肝炎を合併している患者では、本剤の投与中止により、B型慢性肝炎が再燃するおそれがある。特に非代償性の場合、重症化するおそれがある。[1.2参照]
9.1.3 B型又はC型肝炎ウイルス感染患者
9.2 腎機能障害患者
9.2.1 腎機能障害(クレアチニンクリアランスが30mL/min未満)を有する患者
9.2.2 腎機能障害(クレアチニンクリアランスが30〜49mL/min)を有する患者
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。
海外の観察研究において、無脳症や二分脊椎などの神経管閉鎖障害が、受胎前からドルテグラビル含有製剤を服用していた妊婦から生まれた児9460例中10例(0.11%、95%信頼区間0.06-0.19)に報告されており、ドルテグラビルを含まない抗HIV薬を服用していた妊婦から生まれた児23664例中25例(0.11%、95%信頼区間0.07-0.16)、HIV陰性の妊婦から生まれた児170723例中108例(0.07%、95%信頼区間0.05-0.08)に報告されている1)。
海外の観察研究において、無脳症や二分脊椎などの神経管閉鎖障害が、受胎前からドルテグラビル含有製剤を服用していた妊婦から生まれた児9460例中10例(0.11%、95%信頼区間0.06-0.19)に報告されており、ドルテグラビルを含まない抗HIV薬を服用していた妊婦から生まれた児23664例中25例(0.11%、95%信頼区間0.07-0.16)、HIV陰性の妊婦から生まれた児170723例中108例(0.07%、95%信頼区間0.05-0.08)に報告されている1)。
9.5.1 ドルテグラビル
ドルテグラビルはヒト胎盤を通過する。ドルテグラビルの母体血漿中濃度に対する胎児臍帯血漿中濃度の比(中央値[範囲])は、1.28[1.21-1.28]であることが報告されている2)(外国人データ)。
9.5.2 ラミブジン
ヒト胎盤を通過する。出生児の血清中ラミブジン濃度は、分娩時の母親の血清中及び臍帯血中の濃度と同じであることが報告されている(外国人データ)。動物実験(ウサギ)で胎児毒性(早期の胚死亡数の増加)が報告されている。NRTIを子宮内曝露又は周産期曝露された新生児及び乳児において、ミトコンドリア障害によると考えられる軽微で一過性の血清乳酸値の上昇が報告されている。また、非常にまれに発育遅延、てんかん様発作、他の神経疾患も報告されている。しかしながら、これら事象とNRTIの子宮内曝露、周産期曝露との関連性は確立していない。
9.6 授乳婦
本剤投与中は授乳を避けさせること。一般に、HIVの乳児への移行を避けるため、あらゆる状況下においてHIVに感染した女性は授乳すべきでない。
9.6.1 ドルテグラビル
ドルテグラビルはヒト乳汁中に移行する。ドルテグラビルの母体血漿中濃度に対する乳汁中濃度の比(中央値[範囲])は、0.033[0.021-0.050]であることが報告されている2)(外国人データ)。
9.6.2 ラミブジン
経口投与されたラミブジンはヒト乳汁中に排泄されることが報告されている(乳汁中濃度:<0.5-8.2μg/mL)3)(外国人データ)。ラミブジンの母体血漿中濃度に対する乳汁中濃度の比は0.6〜3.3であることが報告されている(外国人データ)。乳児の血清中のラミブジン濃度は18〜28ng/mLであったとの報告がある(外国人データ)。
9.7 小児等
9.7.1 小児等を対象とした本剤の臨床試験は実施していない。
9.8 高齢者
患者の状態を観察しながら注意して投与すること。一般に、高齢者では生理機能(肝機能、腎機能、心機能等)が低下しており、合併症を有している又は他の薬剤を併用している場合が多い。ラミブジンは、主として未変化体として腎から排泄されるが、高齢者では腎機能が低下していることが多いため、高い血中濃度が持続するおそれがある。
10. 相互作用
相互作用序文
薬物代謝酵素用語
UGT1A1
薬物代謝酵素用語
CYP3A4
薬物代謝酵素用語
有機カチオントランスポーター2(OCT2)
薬物代謝酵素用語
Multidrug and Toxin Extrusion 1(MATE1)
薬物代謝酵素用語
MATE2-K
10.2 併用注意
| ピルシカイニド塩酸塩水和物 | ピルシカイニドの血漿中濃度を上昇させる可能性がある。併用により、ピルシカイニドで重大な副作用として報告されている心室頻拍、洞停止及び心室細動等の発現及び重篤化があらわれるおそれがあるので、併用中は注意深く観察すること。 | ドルテグラビルのOCT2及びMATE1の阻害作用により、ピルシカイニドの排出が阻害される可能性がある。 |
| カルバマゼピン [7.1、7.2、16.7.1参照] | ドルテグラビルの血漿中濃度をCmaxで33%、Cτで73%低下させたとの報告がある。 | カルバマゼピンがCYP3A4及びUGT1A1を誘導することにより、ドルテグラビルの代謝が促進される。 |
| フェニトイン ホスフェニトイン フェノバルビタール セイヨウオトギリソウ(St.John's Wort,セント・ジョーンズ・ワート)含有食品 [7.1、7.2参照] | ドルテグラビルの血漿中濃度を低下させる可能性がある。 | これらの薬剤並びにセイヨウオトギリソウがCYP3A4及びUGT1A1を誘導することにより、ドルテグラビルの代謝が促進される。 |
| リファンピシン [7.1、7.2、16.7.1参照] | ドルテグラビルの血漿中濃度をCmaxで43%、Cτで72%低下させたとの報告がある。 | リファンピシンがCYP3A4及びUGT1A1を誘導することにより、ドルテグラビルの代謝が促進される。 |
| 多価カチオン(Mg,Al等)含有製剤 [16.7.1参照] | ドルテグラビルの血漿中濃度をCmaxで72%、C24で74%低下させる。本剤は多価カチオン含有製剤の投与2時間前又は6時間後の投与が推奨される。 | これらの多価カチオンと錯体を形成することにより、ドルテグラビルの吸収が阻害される。 |
| 鉄剤、カルシウム含有製剤(サプリメント等) [16.7.1参照] | ドルテグラビルの血漿中濃度をCmaxで35%、C24で32%低下させる。食事と同時に摂取する場合を除き、本剤は鉄剤、カルシウム含有製剤の投与2時間前又は6時間後の投与が推奨される。 | 鉄、カルシウムと錯体を形成することにより、ドルテグラビルの吸収が阻害される。 |
| メトホルミン塩酸塩 [16.7.1参照] | メトホルミンの血漿中濃度をドルテグラビル50mg1日1回投与時及び1日2回投与時でCmaxでそれぞれ66%及び111%上昇させる。注意深く観察し、必要に応じてメトホルミンを減量する等慎重に投与すること。 | ドルテグラビルのOCT2及びMATE1の阻害作用により、メトホルミンの排出が阻害される可能性がある。 |
| スルファメトキサゾール・トリメトプリム [16.7.2参照] | ラミブジンのAUCが43%増加し、全身クリアランスが30%、腎クリアランスが35%減少したとの報告がある。 | トリメトプリムのOCT2、MATE1及びMATE2-Kの阻害作用により、ラミブジンの腎排泄が阻害されると考えられている。 |
| ソルビトール | 経口ソルビトール溶液(ソルビトールとして3.2g、10.2g、13.4g)とラミブジンの併用により、ラミブジンのAUCが減少した(それぞれ18%、36%、42%減少)との報告がある。 | ソルビトールによりラミブジンの吸収が抑制されると考えられている。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 薬剤性過敏症症候群(頻度不明)
初期症状として発疹、発熱がみられ、さらに肝機能障害、リンパ節腫脹、好酸球増多等を伴う遅発性の重篤な過敏症状があらわれることがある。なお、投与中止後も発疹、発熱、肝機能障害等の症状が再燃あるいは遷延化することがあるので注意すること。
11.1.2 重篤な血液障害
赤芽球癆(頻度不明)、汎血球減少(頻度不明)、貧血(頻度不明)、白血球減少(頻度不明)、好中球減少(頻度不明)、血小板減少(0.1%)[8.6参照]
11.1.3 膵炎(頻度不明)
11.1.4 乳酸アシドーシス(頻度不明)及び脂肪沈着による重度の肝腫大(脂肪肝)(0.1%)
乳酸アシドーシス又は肝毒性が疑われる臨床症状や検査値異常が認められた場合には、本剤の投与を一時中止すること。特に、肝疾患の危険因子を有する患者においては注意すること。ラミブジンを含むヌクレオシド系逆転写酵素阻害薬(NRTI)の単独投与又はこれらの併用療法により、重篤な乳酸アシドーシス(全身倦怠、食欲不振、急な体重減少、胃腸障害、呼吸困難、頻呼吸等)、肝毒性(脂肪沈着による重度の肝腫大、脂肪肝を含む)が、女性に多く報告されている。[8.6参照]
11.1.5 横紋筋融解症(頻度不明)[8.6参照]
11.1.6 ニューロパチー(頻度不明)、錯乱状態(頻度不明)、痙攣(頻度不明)[8.6参照]
11.1.7 心不全(頻度不明)[8.6参照]
11.1.8 肝機能障害(0.1%)、黄疸(頻度不明)
11.2 その他の副作用
| 1%以上 | 1%未満 | 頻度不明 | |
| 免疫系 | 免疫再構築炎症反応症候群 | ||
| 精神・神経系 | 頭痛、不眠症、不安、めまい、傾眠 | 異常な夢、自殺念慮、うつ病 | 自殺企図、錯感覚、末梢神経障害 |
| 消化器 | 悪心、下痢 | 嘔吐、鼓腸、腹痛、上腹部痛 | 腹部不快感 |
| 肝臓 | 肝炎、肝機能検査値異常(AST、ALT等の上昇) | ||
| 皮膚 | そう痒、脱毛 | 発疹 | |
| 全身症状 | 疲労 | 倦怠感、発熱 | |
| 代謝及び栄養障害 | 体脂肪の再分布/蓄積(胸部、体幹部の脂肪増加、末梢部、顔面の脂肪減少、野牛肩、血清脂質増加、血糖増加) | 高乳酸塩血症、アミラーゼ上昇 | |
| 筋骨格 | 関節痛、筋肉痛 | 筋障害 | |
| 臨床検査 | 体重増加、血清クレアチニン増加 | 総ビリルビン増加、クレアチンホスホキナーゼ増加 |
13. 過量投与
13.1 処置
15. その他の注意
15.2 非臨床試験に基づく情報
ラミブジンについては、遺伝毒性試験において弱い染色体異常誘発作用を示したとの報告がある。また、長期のがん原性試験において発がん性を認めなかったとの報告がある。ヒト末梢血リンパ球を用いた染色体異常試験では300μg/mL以上、マウスリンパ腫細胞を用いた遺伝子突然変異試験では2000μg/mL以上で陽性を示した。マウス及びラットを用いた長期のがん原性試験では、臨床用量におけるヒト全身曝露量(AUC)の10倍(マウス)及び58倍(ラット)までの曝露量において、発がん性は認められなかった。
16. 薬物動態
16.1 血中濃度
16.1.1 健康成人
健康成人76例に本剤(ドルテグラビル・ラミブジン50mg・300mg)を空腹時に単回経口投与した時の血漿中ドルテグラビル及びラミブジンの濃度推移を図-1及び図-2に、薬物動態パラメータを表-1及び表-2に示す7)(外国人データ)。
| AUC0-inf(μg・h/mL) | Cmax(μg/mL) | Tmax(h)注1) | t1/2(h) |
| 54.56(32.12) | 2.91(30.55) | 2.50(0.50,6.00) | 14.99(18.23) |
図-1 健康成人に本剤を単回経口投与した時の血漿中ドルテグラビル濃度推移(平均値+標準偏差)
| AUC0-inf(μg・h/mL) | Cmax(μg/mL) | Tmax(h)注1) | t1/2(h) |
| 13.59(17.99) | 3.22(29.30) | 1.00(0.50,3.50) | 18.63(26.85) |
図-2 健康成人に本剤を単回経口投与した時の血漿中ラミブジン濃度推移(平均値+標準偏差)
16.1.2 成人HIV感染症患者
成人HIV感染症患者に本剤(ドルテグラビル・ラミブジン50mg・300mg)を反復経口投与した時の曝露量の推定値(母集団薬物動態解析)を表-3に示す。
| 患者 | 例数 | AUC0-tau(μg・h/mL) | Cmax(μg/mL) | Ctau(μg/mL) | |
| ドルテグラビル | 日本人 | 5 | 63.1(41.9) | 5.61(68.8) | 0.99(153)注1) |
| 外国人 | 356 | 59.2(90.6) | 5.07(83.3) | 1.23(156) | |
| ラミブジン | 日本人 | 5 | 14.5(70.7) | 3.14(97.2) | 0.0668(193) |
| 外国人 | 356 | 14.1(102) | 2.49(90.5) | 0.0893(229)注2) |
16.1.3 本剤投与時と各単剤投与時の曝露量の比較
健康成人76例に本剤(ドルテグラビル・ラミブジン50mg・300mg)とドルテグラビル(50mg)及びラミブジン(300mg)を空腹時に単回経口投与し、単剤併用投与時と本剤投与時の曝露量を比較した。本剤投与時のドルテグラビルのAUC0-t及びCmaxの比(90%信頼区間)は、単剤投与時と比べてそれぞれ1.1578(1.0718,1.2507)及び1.1410(1.0533,1.2361)、ラミブジンのAUC0-t及びCmaxの比は、それぞれ1.0702(1.0464,1.0946)及び1.3176(1.2616,1.3760)であった7)(外国人データ)。
16.2 吸収
16.2.1 食事の影響
健康成人16例に本剤(ドルテグラビル・ラミブジン50mg・300mg)を食後(高脂肪食)に単回経口投与した時、空腹時と比べて高脂肪食摂取後では、ドルテグラビルのAUC0-t及びCmaxはそれぞれ約32%及び約21%増加し、ラミブジンのAUC0-t及びCmaxはそれぞれ約10%及び約32%低下した7)(外国人データ)。
16.2.2 バイオアベイラビリティ
成人HIV感染症患者にラミブジンのカプセル製剤0.25〜8mg/kg注)を単回経口投与した時の生物学的利用率は約82%であった8)(外国人データ)。
16.3 分布
16.3.1 ドルテグラビル
(1)血漿蛋白結合率
In vitroでの、ドルテグラビルのヒト血漿蛋白結合率は約99.3%であった9)。
(2)分布容積
健康成人男性にドルテグラビル20mg(懸濁液)注)を単回経口投与した時の見かけの分布容積は12.5Lであった(外国人データ)。
(3)血球移行性
ヒトでの血液/血漿比(平均値)は0.441〜0.535であり、ドルテグラビルの血球移行性は低かった(5%未満)。
(4)非結合型薬物
血漿中ドルテグラビルの遊離分画は健康成人で約0.2〜1.1%、中等度の肝機能障害患者で約0.4〜0.5%、重度の腎機能障害患者で約0.8〜1.0%、成人HIV感染症患者で0.5%であった(外国人データ)。
(5)脳脊髄液への移行
ドルテグラビルは脳脊髄液中にも分布する。ドルテグラビル50mg及びアバカビル・ラミブジン(600mg・300mg)が併用投与された抗HIV薬による治療経験のない成人HIV感染症患者11例において、ドルテグラビルの脳脊髄液中濃度(中央値)は18ng/mLであり、血漿中濃度の0.11〜0.66%であった(外国人データ)。
(6)組織内分布
ドルテグラビルは女性及び男性の生殖器に分布する。健康成人女性にドルテグラビル50mg/日を5〜7日間経口投与した時の子宮頸膣液、子宮頸部組織及び膣組織におけるドルテグラビルのAUCは定常状態での血漿中ドルテグラビルのAUCの6〜10%であった(外国人データ)。また、健康成人男性にドルテグラビル50mg/日を8日間経口投与した時の精液及び直腸組織におけるドルテグラビルAUCは定常状態での血漿中ドルテグラビルのAUCの7及び17%であった(外国人データ)。
16.3.2 ラミブジン
成人HIV感染症患者に4〜10mg/kg注)を1日2回2週間以上反復経口投与した時、投与2時間後の脳脊髄液中濃度は血中濃度の約6%であった10)(外国人データ)。
16.4 代謝
16.4.1 ドルテグラビル
(1)代謝酵素
16.4.2 ラミブジン
ヒトでの主代謝物はトランス−スルホキシド体(1-[(2R,5S)-trans-2-hydroxymethyl-1,3-oxathiolan-3-oxide-5-yl]cytosine)であった13)。
16.5 排泄
16.5.1 ドルテグラビル
健康成人にドルテグラビル20mg注)を単回経口投与した時の主な排泄経路は糞であり、経口投与量の53%が未変化体として糞中に排泄された。また、尿中には経口投与量の31%が排泄され、その内訳は18.9%がエーテル型グルクロン酸抱合体、3.6%がN-脱アルキル体、3.0%がベンジル位の酸化体であり、未変化体は1%未満であった(外国人データ)。健康成人に14C-ドルテグラビル20mg(懸濁液)注)を単回経口投与した時の総投与量の約9.7%が酸化的代謝物として尿糞中に回収された(外国人データ)。
16.5.2 ラミブジン
成人HIV感染症患者にラミブジン2mg/kg注)を経口投与した時、投与後12時間尿中にトランス−スルホキシド体が投与量の5.2%排泄された。また、血中濃度が定常状態での未変化体の尿中排泄率は投与量の約70%であり、腎排泄がラミブジンの体内からの除去の主要な経路であることが示された13)(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
(1)ドルテグラビル
重度の腎機能障害(8例、クレアチニンクリアランス:30mL/min未満)を有する患者にドルテグラビル50mgを単回経口投与した時の血漿中ドルテグラビルの薬物動態パラメータを表-4に示す(外国人データ)。重度の腎機能障害患者における薬物動態は健康成人との間に臨床的に重要である差はみられなかったことから、腎機能障害患者に対してドルテグラビルの用量調節を行う必要はない14)。[5.3参照]
| 被験者 | Cmax(μg/mL) | AUC0-inf(μg・h/mL) | t1/2(h) |
| 重度の腎機能障害患者 | 1.50(34) | 23.5(48) | 12.7(31) |
| 健康成人 | 1.86(45) | 37.1(58) | 15.4(15) |
(2)ラミブジン
16.6.2 肝機能障害患者
(1)ドルテグラビル
ドルテグラビルは主に肝臓で代謝されて排泄される。中等度の肝機能障害(8例、Child-Pugh分類:B)を有する患者にドルテグラビル50mgを単回経口投与した時の血漿中ドルテグラビルの薬物動態パラメータを表-5に示す(外国人データ)。中等度の肝機能障害患者における薬物動態は健康成人と同様であったことから、中等度の肝機能障害に対してドルテグラビルの用量調節の必要はない16)。なお、重度の肝機能障害患者でのドルテグラビルの薬物動態に及ぼす影響については検討していない。
| 被験者 | AUC0-inf(μg・h/mL) | Cmax(μg/mL) | C24(μg/mL) |
| 中等度の肝機能障害患者 | 38.5(30) | 1.78(17) | 0.59(36) |
| 健康成人 | 37.3(47) | 1.80(49) | 0.57(44) |
(2)ラミブジン
中等度及び重度の肝障害を有する患者における成績より、ラミブジンの薬物動態は、肝障害によって重大な影響を受けないことが示されている17)。
16.6.3 小児
(1)ドルテグラビル
抗HIV薬による治療経験のある小児HIV感染症患者(12歳以上18歳未満、10例)にドルテグラビル50mgを1日1回経口投与した時の薬物動態は成人と同様であった。小児患者での血漿中ドルテグラビルの薬物動態パラメータを表-6に示す(外国人データ)。
| 年齢/体重 | 用量 | 薬物動態パラメータの推定値 | ||
| AUC0-24(μg・h/mL) | Cmax(μg/mL) | C24(μg/mL) | ||
| 12歳以上18歳未満 体重40kg以上注1) | 50mg注1) 1日1回 | 46(43) | 3.49(38) | 0.90(59) |
(2)ラミブジン
12歳以上の小児HIV-1感染症患者(114例)にラミブジン300mg/日(150mg1日2回又は300mg1日1回)を反復経口投与した時のPKパラメータは表-7のとおりであった(外国人データ)18)。
| 用法・用量 | AUC0-24(μg・h/mL) | Cmax(μg/mL) | Cmin(μg/mL) |
| 150mg 1日2回 | 14.8(3.8) | 1.2(0.32) | 0.15(0.09) |
| 300mg 1日1回 | 12.8(3.6) | 1.9(0.44) | 0.07(0.06) |
小児及び成人のHIV感染症患者を対象にラミブジンを投与した時、年齢とCL/Fの関係は図-3のとおりであり、成人と12歳以上の小児においてラミブジンのCL/Fは同程度であった。12歳以上の小児患者及び成人患者におけるラミブジンの曝露量に臨床的に意味のある差異は認められなかった。
図-3 小児及び成人HIV-1感染症患者におけるラミブジンのCL/F(L/hr)と年齢の関係
小児HIV感染症患者に4mg/kg注)を単回経口投与した時、生物学的利用率は約66%であり、成人HIV感染症患者の生物学的利用率(約82%)より低い値を示した。小児で生物学的利用率が減少する機序は不明である。また、脳脊髄液中のラミブジンの濃度は血中濃度の約13%であった(外国人データ)13)。
注)本剤の承認された用法及び用量は「通常、成人及び12歳以上かつ体重40kg以上の小児には、1回1錠(ドルテグラビルとして50mg及びラミブジンとして300mg)を食事の有無に関わらず1日1回経口投与する。」である。
16.7 薬物相互作用
16.7.1 ドルテグラビル
(1)In vitro
(2)In vivo
ドルテグラビル製剤を併用薬と投与した時の薬物動態パラメータの変化を表-8及び表-9に示す(外国人データ)。[7.2、10.2参照]
| 併用薬及び用量 | ドルテグラビルの用量 | 例数 | ドルテグラビル併用時/非併用時の併用薬の薬物動態パラメータの幾何平均の比(90%信頼区間) | ||
| Ctau又はC24 | AUC | Cmax | |||
| エチニルエストラジオール0.035mg23) | 50mg 1日2回 | 15 | 1.02(0.93,1.11) | 1.03(0.96,1.11) | 0.99(0.91,1.08) |
| メサドン 20-150mg24) | 50mg 1日2回 | 11 | 0.99(0.91,1.07) | 0.98(0.91,1.06) | 1.00(0.94,1.06) |
| ミダゾラム3mg25) | 25mg 1日1回 | 10 | − | 0.95(0.79,1.15) | − |
| Norelgestromin(国内未発売) 0.25mg23) | 50mg 1日2回 | 15 | 0.93(0.85,1.03) | 0.98(0.91,1.04) | 0.89(0.82,0.97) |
| テノホビル ジソプロキシルフマル酸塩 300mg 1日1回26) | 50mg 1日1回 | 15 | 1.19(1.04,1.35) | 1.12(1.01,1.24) | 1.09(0.97,1.23) |
| メトホルミン 500mg 1日2回27) | 50mg 1日1回 | 14 | − | 1.79(1.65,1.93) | 1.66(1.53,1.81) |
| メトホルミン 500mg 1日2回27) | 50mg 1日2回 | 14 | − | 2.45(2.25,2.66) | 2.11(1.91,2.33) |
| ダクラタスビル 60mg 1日1回28) | 50mg 1日1回 | 12 | 1.06(0.88,1.29) | 0.98(0.83,1.15) | 1.03(0.84,1.25) |
| 併用薬及び用量 | ドルテグラビルの用量 | 例数 | 他剤併用時/非併用時のドルテグラビルの薬物動態パラメータの幾何平均の比(90%信頼区間) | ||
| Ctau又はC24 | AUC | Cmax | |||
| アタザナビル 400mg 1日1回29) | 30mg 1日1回 | 12 | 2.80(2.52,3.11) | 1.91(1.80,2.03) | 1.50(1.40,1.59) |
| アタザナビル+リトナビル 300mg+100mg 1日1回29) | 30mg 1日1回 | 12 | 2.21(1.97,2.47) | 1.62(1.50,1.74) | 1.34(1.25,1.42) |
| テノホビル ジソプロキシルフマル酸塩 300mg 1日1回26) | 50mg 1日1回 | 15 | 0.92(0.82,1.04) | 1.01(0.91,1.11) | 0.97(0.87,1.08) |
| ダルナビル+リトナビル 600mg+100mg 1日2回30) | 30mg 1日1回 | 15 | 0.62(0.56,0.69) | 0.78(0.72,0.85) | 0.89(0.83,0.97) |
| エファビレンツ 600mg 1日1回31) | 50mg 1日1回 | 12 | 0.25(0.18,0.34) | 0.43(0.35,0.54) | 0.61(0.51,0.73) |
| エトラビリン 200mg 1日2回32) | 50mg 1日1回 | 15 | 0.12(0.09,0.16) | 0.29(0.26,0.34) | 0.48(0.43,0.54) |
| エトラビリン+ダルナビル+リトナビル 200mg+600mg+100mg 1日2回33) | 50mg 1日1回 | 9 | 0.63(0.52,0.76) | 0.75(0.69,0.81) | 0.88(0.78,1.00) |
| エトラビリン+ロピナビル・リトナビル 200mg+400mg・100mg 1日2回33) | 50mg 1日1回 | 8 | 1.28(1.13,1.45) | 1.11(1.02,1.20) | 1.07(1.02,1.13) |
| ホスアンプレナビル+リトナビル 700mg+100mg 1日2回34) | 50mg 1日1回 | 12 | 0.51(0.41,0.63) | 0.65(0.54,0.78) | 0.76(0.63,0.92) |
| ロピナビル・リトナビル 400mg・100mg 1日2回30) | 30mg 1日1回 | 15 | 0.94(0.85,1.05) | 0.97(0.91,1.04) | 1.00(0.94,1.07) |
| 乾燥水酸化アルミニウムゲル・水酸化マグネシウム 20mL 単回35) | 50mg 単回 | 16 | 0.26(0.21,0.31) | 0.26(0.22,0.32) | 0.28(0.23,0.33) |
| 乾燥水酸化アルミニウムゲル・水酸化マグネシウム 20mL 投与後2時間 単回35) | 50mg 単回 | 16 | 0.70(0.58,0.85) | 0.74(0.62,0.90) | 0.82(0.69,0.98) |
| 総合ビタミン剤 1錠 1日1回35) | 50mg 単回 | 16 | 0.68(0.56,0.82) | 0.67(0.55,0.81) | 0.65(0.54,0.77) |
| 炭酸カルシウム 1200mg 単回(空腹時)36) | 50mg 単回 | 12 | 0.61(0.47,0.80) | 0.61(0.47,0.80) | 0.63(0.50,0.81) |
| 炭酸カルシウム 1200mg 単回(食後)36) | 50mg 単回 | 11 | 1.08(0.81,1.42) | 1.09(0.84,1.43) | 1.07(0.83,1.38) |
| 炭酸カルシウム 1200mg 投与後2時間 単回36) | 50mg 単回 | 11 | 0.90(0.68,1.19) | 0.94(0.72,1.23) | 1.00(0.78,1.29) |
| フマル酸第一鉄 324mg 単回(空腹時)36) | 50mg 単回 | 11 | 0.44(0.36,0.54) | 0.46(0.38,0.56) | 0.43(0.35,0.52) |
| フマル酸第一鉄 324mg 単回(食後)36) | 50mg 単回 | 10 | 1.00(0.81,1.23) | 0.98(0.81,1.20) | 1.03(0.84,1.26) |
| フマル酸第一鉄 324mg 投与後2時間 単回36) | 50mg 単回 | 10 | 0.92(0.74,1.13) | 0.95(0.77,1.15) | 0.99(0.81,1.21) |
| オメプラゾール 40mg 1日1回37) | 50mg 単回 | 12 | 0.95(0.75,1.21) | 0.97(0.78,1.20) | 0.92(0.75,1.11) |
| prednisone(国内未発売) 60mg 1日1回(漸減)38) | 50mg 1日1回 | 12 | 1.17(1.06,1.28) | 1.11(1.03,1.20) | 1.06(0.99,1.14) |
| リファンピシン注1) 600mg 1日1回39) | 50mg 1日2回注1) | 11 | 0.28(0.23,0.34) | 0.46(0.38,0.55) | 0.57(0.49,0.65) |
| リファンピシン注2) 600mg 1日1回39) | 50mg 1日2回注2) | 11 | 1.22(1.01,1.48) | 1.33(1.15,1.53) | 1.18(1.03,1.37) |
| リファブチン 300mg 1日1回39) | 50mg 1日1回 | 9 | 0.70(0.57,0.87) | 0.95(0.82,1.10) | 1.16(0.98,1.37) |
| Tipranavir(国内未発売)+リトナビル 500mg+200mg 1日2回40) | 50mg 1日1回 | 14 | 0.24(0.21,0.27) | 0.41(0.38,0.44) | 0.54(0.50,0.57) |
| テラプレビル 750mg 8時間毎41) | 50mg 1日1回 | 15 | 1.37(1.29,1.45) | 1.25(1.20,1.31) | 1.19(1.11,1.26) |
| Boceprevir(国内未発売) 800mg 8時間毎41) | 50mg 1日1回 | 13 | 1.08(0.91,1.28) | 1.07(0.95,1.20) | 1.05(0.96,1.15) |
| カルバマゼピン 300mg 1日2回42) | 50mg 1日1回 | 14 | 0.27(0.24,0.31) | 0.51(0.48,0.55) | 0.67(0.61,0.73) |
| ダクラタスビル 60mg 1日1回28) | 50mg 1日1回 | 12 | 1.45(1.25,1.68) | 1.33(1.11,1.59) | 1.29(1.07,1.57) |
16.7.2 ラミブジン
ラミブジンの薬物動態に及ぼす併用薬の影響を表-10に示す(外国人データ)。[10.2参照]
| 併用薬及び用量 | ラミブジンの用量 | 例数 | 他剤併用時/非併用時のラミブジンの薬物動態パラメータの幾何平均比(90%信頼区間);影響なし=1.00 | ||
| CL/F | AUC | CLr | |||
| トリメトプリム・スルファメトキサゾール 160mg・800mg/日 5日間 | ラミブジン300mg 単回 | 14 | 0.70(0.65,0.76) | 1.43(1.32,1.55) | 0.65(0.54,0.78) |
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 海外第III相試験(GEMINI-1:204861試験)
主要な耐性変異(IAS-USA定義)45)を有さず、抗HIV薬による治療経験のない成人HIV-1感染症患者714例を対象とした二重盲検比較試験において、ドルテグラビル(50mg)とラミブジン(300mg)の1日1回併用投与群(DTG+3TC群)に356例、ドルテグラビルとテノホビルジソプロキシルフマル酸塩・エムトリシタビン配合剤の1日1回併用投与群(DTG+TDF・FTC群)に358例が無作為に割り付けられた。その結果、主要評価項目である投与48週時のHIV-1 RNA量が50copies/mL未満であった被験者の割合は、DTG+3TC群の90%に対して、DTG+TDF・FTC群は93%であり、調整した群間差の95%信頼区間の下限値(−6.7%)は、非劣性マージン(−10%)より大きく、DTG+TDF・FTCレジメンに対するDTG+3TCレジメンの非劣性が検証された。[5.1参照]
なお、本試験における試験成績の要約を表-1に示した。
なお、本試験における試験成績の要約を表-1に示した。
| DTG+3TC群 356例 | DTG+TDF・FTC群 358例 | |
| 48週 | 48週 | |
| HIV-1 RNA量が50copies/mL未満 | 320例(90%) | 332例(93%) |
| 両群間の差(95%信頼区間)注1) | −2.6%(−6.7%,1.5%) | |
| ウイルス学的な治療失敗注2) | 13例(4%) | 6例(2%) |
| DTG+3TC群356例 | DTG+TDF・FTC群358例 | 割合の差とその95%信頼区間注1) | ||
| 48週 | 48週 | |||
| HIV-1 RNA量 | 100,000copies/mL以下 | 255/282例(90%) | 263/282例(93%) | −2.8%(−7.3%,1.7%) |
| 100,000copies/mL超 | 65/74例(88%) | 69/76例(91%) | −3.0%(−12.8%,6.9%) | |
| CD4陽性細胞数 | 200cell/mm3以下 | 25/31例(81%) | 26/29例(90%) | −9.0%(−26.8%,8.8%) |
| 200cell/mm3超 | 295/325例(91%) | 306/329例(93%) | −2.2%(−6.4%,1.9%) | |
副作用発現頻度は、DTG+3TC群で20%(71/356例)であった。主な副作用は、頭痛4%(14/356例)、悪心2%(8/356例)、不眠症2%(7/356例)、傾眠2%(7/356例)、下痢2%(6/356例)及び疲労2%(6/356例)であった。(投与48週時)
17.1.2 海外第III相試験(GEMINI-2:205543試験)
主要な耐性変異(IAS-USA定義)45)を有さず、抗HIV薬による治療経験のない成人HIV-1感染症患者719例を対象とした二重盲検比較試験において、ドルテグラビル(50mg)とラミブジン(300mg)の1日1回併用投与群(DTG+3TC群)に360例、ドルテグラビルとテノホビルジソプロキシルフマル酸塩・エムトリシタビン配合剤の1日1回併用投与群(DTG+TDF・FTC群)に359例が無作為に割り付けられた。その結果、主要評価項目である投与48週時のHIV-1 RNA量が50copies/mL未満であった被験者の割合は、DTG+3TC群の93%に対して、DTG+TDF・FTC群は94%であり、調整した群間差の95%信頼区間の下限値(−4.3%)は、非劣性マージン(−10%)より大きく、DTG+TDF・FTCレジメンに対するDTG+3TCレジメンの非劣性が検証された。[5.1参照]
なお、本試験における試験成績の要約を表-3に示した。
なお、本試験における試験成績の要約を表-3に示した。
| DTG+3TC群 360例 | DTG+TDF・FTC群 359例 | |
| 48週 | 48週 | |
| HIV-1 RNA量が50copies/mL未満 | 335例(93%) | 337例(94%) |
| 両群間の差(95%信頼区間)注1) | −0.7%(−4.3%,2.9%) | |
| ウイルス学的な治療失敗注2) | 7例(2%) | 7例(2%) |
| DTG+3TC群360例 | DTG+TDF・FTC群359例 | 割合の差とその95%信頼区間注1) | ||
| 48週 | 48週 | |||
| HIV-1 RNA量 | 100,000copies/mL以下 | 271/294例(92%) | 268/282例(95%) | −2.9%(−6.8%,1.1%) |
| 100,000copies/mL超 | 64/66例(97%) | 69/77例(90%) | 7.4%(−0.6%,15.3%) | |
| CD4陽性細胞数 | 200cell/mm3以下 | 25/32例(78%) | 25/26例(96%) | −18.0%(−34.1%,−1.9%) |
| 200cell/mm3超 | 310/328例(95%) | 312/333例(94%) | 0.8%(−2.8%,4.4%) | |
副作用発現頻度は、DTG+3TC群で15%(55/360例)であった。主な副作用は、下痢2%(8/360例)、頭痛2%(7/360例)、悪心2%(6/360例)及び不眠症2%(6/360例)であった。(投与48週時)
17.1.3 国際共同第III相試験(TANGO:204862試験)
主要なNRTI耐性変異又は主要なINSTI耐性変異(IAS-USA定義)45)を有さず、抗レトロウイルス療法(TAF・FTCとINSTI、NNRTI又はPIのいずれか1剤)によりウイルス学的に抑制されている成人HIV-1感染症患者741例(日本人患者11例を含む)を対象とした非盲検比較試験において、本剤1日1回投与群に369例、現行のレジメンを継続する群(継続投与群)に372例が無作為に割り付けられた。その結果、主要評価項目である投与48週時のウイルス学的な治療失敗であった被験者の割合は、本剤群の0.3%に対して、継続投与群は0.5%であり、調整した群間差の95%信頼区間の上限値(0.7%)は、非劣性マージン(4%)より小さく、継続投与レジメンに対する本剤の非劣性が検証された。[5.1参照]
なお、日本人11例(本剤群5例、継続投与群6例)における48週時のウイルス学的な治療失敗であった被験者の割合は、本剤群及び継続投与群ともに0%であった。
本試験における試験成績の要約を表-5に示した。
なお、日本人11例(本剤群5例、継続投与群6例)における48週時のウイルス学的な治療失敗であった被験者の割合は、本剤群及び継続投与群ともに0%であった。
本試験における試験成績の要約を表-5に示した。
| 本剤群 369例 | 継続投与群 372例 | |
| 48週 | 48週 | |
| ウイルス学的な治療失敗注1) | 1例(0.3%) | 2例(0.5%) |
| 両群間の差(95%信頼区間)注2) | −0.3%(−1.2%,0.7%) | |
| HIV-1 RNA量が50copies/mL未満 | 344例(93.2%) | 346例(93.0%) |
副作用発現頻度は、本剤群で12%(45/369例)であった。主な副作用は、不眠症2%(6/369例)、悪心1%(5/369例)、下痢1%(4/369例)及び不安1%(4/369例)であった。(投与48週時)
18. 薬効薬理
18.1 作用機序
18.1.1 ドルテグラビル
ドルテグラビルはレトロウイルスの複製に必要な酵素であるHIVインテグラーゼの活性部位に結合することによってその活性を阻害し、ウイルスDNAの宿主DNAへの組込みを抑制する。
18.1.2 ラミブジン
ラミブジンは細胞内でリン酸化され、HIVを感染させた細胞内での半減期が約12時間の活性化型の三リン酸化体に変換される。ラミブジン三リン酸化体はHIVの逆転写酵素によりデオキシシチジン三リン酸の代わりにウイルスDNA鎖に取り込まれ、DNA鎖の伸長を停止させることによりHIVの複製を阻害する。また、ラミブジン三リン酸化体はHIVの逆転写酵素を競合的に阻害する。一方、in vitroで、ヒト末梢血リンパ球、リンパ球系・単球−マクロファージ系の株化細胞及び種々のヒト骨髄前駆細胞に対するラミブジンの細胞毒性は弱かった。
18.2 抗ウイルス作用
18.2.1 ドルテグラビル
HIV-1 BaL株及びHIV-1 NL432株に感染させた末梢血単核球を用いた時のドルテグラビルのウイルス複製に対する50%阻害濃度(IC50)は、それぞれ0.51及び0.53nMであり、HIV-1 IIIB株に感染させたMT-4細胞を用いた時のIC50は2.1nMであった(in vitro)。
13種のHIV-1臨床分離株(サブタイプB)のインテグラーゼコード領域を導入した組換えウイルスに対するドルテグラビルのIC50(平均値)は0.52nMであり、その活性は実験室株に対する抗ウイルス活性と同程度であった。24種のHIV-1臨床分離株[グループM(サブタイプA、B、C、D、E、F、G)及びグループO]並びに3種のHIV-2臨床分離株からなるパネル株を感染させた末梢血単核球を用いた時のドルテグラビルのIC50(幾何平均)はHIV-1株及びHIV-2株でそれぞれ0.20nM(範囲は0.02〜2.14nM)及び0.18nM(範囲は0.09〜0.61nM)であった(in vitro)。
13種のHIV-1臨床分離株(サブタイプB)のインテグラーゼコード領域を導入した組換えウイルスに対するドルテグラビルのIC50(平均値)は0.52nMであり、その活性は実験室株に対する抗ウイルス活性と同程度であった。24種のHIV-1臨床分離株[グループM(サブタイプA、B、C、D、E、F、G)及びグループO]並びに3種のHIV-2臨床分離株からなるパネル株を感染させた末梢血単核球を用いた時のドルテグラビルのIC50(幾何平均)はHIV-1株及びHIV-2株でそれぞれ0.20nM(範囲は0.02〜2.14nM)及び0.18nM(範囲は0.09〜0.61nM)であった(in vitro)。
18.2.2 ラミブジン
In vitroでのラミブジンのHIV-1(RF、GB8、U455及びIIIB株)に対するIC50は670nM以下であり、HIV-2 ROD株に対するIC50は40nMであった。
In vitroでの26種のHIV-1臨床分離株[グループM(サブタイプA、B、C、D、E、F、G)]並びに3種類のHIV-2臨床分離株に対するラミブジンのIC50(平均値)はHIV-1株及びHIV-2株でそれぞれ40nM(範囲は1〜120nM)及び42nM(範囲は2〜120nM)であった。
In vitroでの26種のHIV-1臨床分離株[グループM(サブタイプA、B、C、D、E、F、G)]並びに3種類のHIV-2臨床分離株に対するラミブジンのIC50(平均値)はHIV-1株及びHIV-2株でそれぞれ40nM(範囲は1〜120nM)及び42nM(範囲は2〜120nM)であった。
18.3 薬剤耐性
18.3.1 ドルテグラビル
HIV-1 IIIB株及びHIV-1 NL432株をそれぞれ112及び56日間継代培養した試験でみられたインテグラーゼ領域のアミノ酸変異はS153Y、S153F、E92Q及びG193Eであり、FC(各種分離株に対するIC50/野生型HIV-1株に対するIC50)の最大値は4.1であった。また、HIV-1臨床分離株(サブタイプB、C及びA/G)を更に長期間継代培養した試験でみられた変異はG118R(FC=10)、S153T及びR263K(FC=1.5)であった(in vitro)。
18.3.2 ラミブジン
ラミブジンを含む抗HIV薬で治療を受けたHIV-1感染症患者で発現するラミブジン耐性HIV-1には、HIV逆転写酵素の活性部位に近い逆転写酵素領域の184番目のアミノ酸のメチオニンからバリンへの変異(M184V)がみられる。このM184V変異の結果、ウイルスのラミブジンに対する感受性は著明に低下し、in vitroでのウイルスの複製能力は低下する。
18.3.3 ドルテグラビル及びラミブジン併用
抗HIV薬による治療経験のないHIV-1感染症患者
海外第III相試験(GEMINI-1:204861試験及びGEMINI-2:205543試験)において、投与48週までにウイルス学的中止基準を満たした症例は全体で10例(ドルテグラビル及びラミブジン併用投与群で6例)であったが、いずれの被験者においてもINSTI及びNRTI耐性変異は認められなかった。
18.4 交差耐性
18.4.1 ドルテグラビル
部位特異的変異を有する60種のINSTI耐性HIV-1ウイルスパネル株(28種は単一アミノ酸変異、32種は二重又は多重変異)を用いてドルテグラビルの抗ウイルス活性を検討した。単一のINSTI耐性関連アミノ酸変異(T66K、I151L及びS153Y)を有するウイルスでは、ドルテグラビルに対する感受性が2倍以上(2.3〜3.6倍)低下した。複数の変異(T66K/L74M、E92Q/N155H、G140C/Q148R、G140S/Q148H、G140S/Q148R、G140S/Q148K、Q148R/N155H、T97A/G140S/Q148、及びE138/G140/Q148)を有するウイルスでは、ドルテグラビルに対する感受性が2倍以上(2.5〜21倍)低下した(in vitro)。705種のラルテグラビル耐性臨床分離株のうち93.9%の株に対するFCは10以下であった(in vitro)。
18.4.2 ラミブジン
ジドブジン及びサニルブジンは、ラミブジン耐性HIV-1に対し抗ウイルス活性を維持する。アバカビルは逆転写酵素領域のM184V変異のみが認められているウイルスに対しては、抗ウイルス活性を維持する。また、ジダノシン及びザルシタビンは、M184V変異ウイルスに対して感受性が低下するというin vitroでの報告があるが、これらの感受性の低下と臨床効果の関係は明らかにされていない。
19. 有効成分に関する理化学的知見
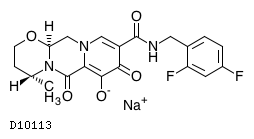
19.1. ドルテグラビルナトリウム
| 一般的名称 | ドルテグラビルナトリウム |
|---|---|
| 一般的名称(欧名) | Dolutegravir Sodium |
| 化学名 | Monosodium(4R,12aS)-9-{[(2,4-difluorophenyl)methyl]carbamoyl}-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1',2':4,5]pyrazino[2,1-b][1,3]oxazin-7-olate |
| 分子式 | C20H18F2N3NaO5 |
| 分子量 | 441.36 |
| 融点 | 1型結晶は約350℃で溶融と同時に分解する。 |
| 物理化学的性状 | 白色〜淡黄白色の粉末。水に溶けにくく、エタノール(99.5)にほとんど溶けない。 |
| 分配係数 | (logP):2.16±0.01(23℃) |
| KEGG DRUG |
|
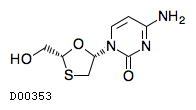
19.2. ラミブジン
| 一般的名称 | ラミブジン |
|---|---|
| 一般的名称(欧名) | Lamivudine |
| 化学名 | 1-[(2R,5S)-2-hydroxymethyl-1,3-oxathiolan-5-yl]cytosine |
| 分子式 | C8H11N3O3S |
| 分子量 | 229.26 |
| 融点 | 約176℃ |
| 物理化学的性状 | 白色〜微黄白色の結晶性の粉末である。ジメチルスルホキシドに溶けやすく、水にやや溶けやすく、メタノール又はエタノール(99.5)にやや溶けにくく、ジエチルエーテルにほとんど溶けない。 |
| 分配係数 | (logP):−0.9(1-オクタノール/水系) |
| KEGG DRUG |
|
21. 承認条件
21.1 医薬品リスク管理計画を策定の上、適切に実施すること。
21.2 本剤の使用に当たっては、患者に対して本剤に関して更なる有効性・安全性のデータを引き続き収集中であること等を十分に説明し、インフォームドコンセントを得るよう、医師に要請すること。
21.3 海外において現在実施中又は計画中の臨床試験については、終了後速やかに試験成績及び解析結果を提出すること。
21.4 再審査期間が終了するまでの間、原則として国内の全投与症例を対象とした製造販売後調査を実施し、本剤の使用実態に関する情報(患者背景、有効性・安全性(他剤併用時の有効性・安全性を含む)及び薬物相互作用のデータ等)を収集して定期的に報告するとともに、調査の結果を再審査申請時に提出すること。
22. 包装
30錠[瓶、バラ]
23. 主要文献
- Zash R,et al., International AIDS Conference 2022., (Poster PELBB02)
- Dickinson L,et al., Clin Infect Dis., 73, e1200-e1207, (2021) »PubMed
- Moodley J,et al., J Infect Dis., 178, 1327-1333, (1998) »PubMed »DOI
- Molto J,et al., Antimicrob Agents Chemother., 60 (4), 2564-2566, (2016) »PubMed
- Bollen P,et al., AIDS., 30, 1490-1491, (2016) »PubMed
- Johnson MA,et al., Br J Clin Pharmacol., 46, 21-27, (1998) »PubMed
- 社内資料:海外臨床試験(204994)
- van Leeuwen R,et al., AIDS., 6, 1471-1475, (1992) »PubMed »DOI
- 社内資料:分布に関する試験(2011N119355、テビケイ錠50mg 2014年3月24日承認、CTD 2.4.3.3.3.1.)
- van Leeuwen R,et al., J Infect Dis., 171, 1166-1171, (1995) »PubMed »DOI
- 社内資料:代謝に関する試験(RD2008/01339、テビケイ錠50mg 2014年3月24日承認、CTD 2.4.3.3.6.1.)
- 社内資料:代謝に関する試験(RD2008/00373、テビケイ錠50mg 2014年3月24日承認、CTD 2.4.3.3.6.1.)
- Johnson MA,et al., Clin Pharmacokinet., 36, 41-66, (1999) »PubMed
- Weller S,et al., Eur J Clin Pharmacol., 70 (1), 29-35, (2014) »PubMed
- Heald AE,et al., Antimicrob Agents Chemother., 40, 1514-1519, (1996) »PubMed »DOI
- 社内資料:海外臨床試験(ING113097、テビケイ錠50mg 2014年3月24日承認、CTD 2.7.2.2.1.2.2.)
- Johnson MA,et al., Eur J Clin Pharmacol., 54, 363-366, (1998) »PubMed »DOI
- Bouazza N,et al., Antimicrob Agents Chemother., 55, 3498-3504, (2011) »PubMed
- 社内資料:分布に関する試験(RD2008/00361、テビケイ錠50mg 2014年3月24日承認、CTD 2.4.3.3.3.2.)
- 社内資料:分布に関する試験(2011N112380、テビケイ錠50mg 2014年3月24日承認、CTD 2.4.3.3.3.2.)
- 社内資料:排泄に関する試験(2010N104937、テビケイ錠50mg 2014年3月24日承認、CTD 2.4.3.3.6.2.)
- 社内資料:排泄に関する試験(2013N161621、テビケイ錠50mg 2014年3月24日承認、CTD 2.4.3.3.6.2.)
- 社内資料:海外臨床試験(ING111855、テビケイ錠50mg 2014年3月24日承認、CTD 2.7.2.2.1.3.7.)
- 社内資料:海外臨床試験(ING115698、テビケイ錠50mg 2014年3月24日承認、CTD 2.7.2.2.1.3.16.)
- 社内資料:海外臨床試験(ING111322、テビケイ錠50mg 2014年3月24日承認、CTD 2.7.2.2.1.3.1.)
- 社内資料:海外臨床試験(ING111604、テビケイ錠50mg 2014年3月24日承認、CTD 2.7.2.2.1.3.5.)
- 社内資料:海外臨床試験(201167)
- Ross LL,et al., BMC Infect Dis., 16, 347, (2016) »PubMed
- 社内資料:海外臨床試験(ING111854、テビケイ錠50mg 2014年3月24日承認、CTD 2.7.2.2.1.3.6.)
- 社内資料:海外臨床試験(ING111405、テビケイ錠50mg 2014年3月24日承認、CTD 2.7.2.2.1.3.2.)
- 社内資料:海外臨床試験(ING114005、テビケイ錠50mg 2014年3月24日承認、CTD 2.7.2.2.1.3.13.)
- 社内資料:海外臨床試験(ING111603、テビケイ錠50mg 2014年3月24日承認、CTD 2.7.2.2.1.3.4.)
- 社内資料:海外臨床試験(ING112934、テビケイ錠50mg 2014年3月24日承認、CTD 2.7.2.2.1.3.8.)
- 社内資料:海外臨床試験(ING113068、テビケイ錠50mg 2014年3月24日承認、CTD 2.7.2.2.1.3.10.)
- 社内資料:海外臨床試験(ING111602、テビケイ錠50mg 2014年3月24日承認、CTD 2.7.2.2.1.3.3.)
- Song I,et al., J Clin Pharmacol., 55 (5), 490-496, (2015) »PubMed
- 社内資料:海外臨床試験(ING112941、テビケイ錠50mg 2014年3月24日承認、CTD 2.7.2.2.1.3.9.)
- 社内資料:海外臨床試験(ING115696、テビケイ錠50mg 2014年3月24日承認、CTD 2.7.2.2.1.3.14.)
- 社内資料:海外臨床試験(ING113099、テビケイ錠50mg 2014年3月24日承認、テビケイ CTD 2.7.2.2.1.3.12.)
- 社内資料:海外臨床試験(ING113096、テビケイ錠50mg 2014年3月24日承認、CTD 2.7.2.2.1.3.11.)
- 社内資料:海外臨床試験(ING115697、テビケイ錠50mg 2014年3月24日承認、CTD 2.7.2.2.1.3.15.)
- Song I,et al., Eur J Clin Pharmacol., 72, 665-670, (2016) »PubMed
- Jung N,et al., Drug Metab Dispos., 36, 1616-1623, (2008) »PubMed
- Muller F,et al., Biochem Pharmacol., 86, 808-815, (2013) »PubMed
- Wensing AM,et al., Top Antivir Med., 24 (4), 132-141, (2016) »PubMed
24. 文献請求先及び問い合わせ先
文献請求先
グラクソ・スミスクライン株式会社
ヴィーブヘルスケア・カスタマー・サービス
東京都港区赤坂1-8-1
電話:0120-066-525 (9:00〜17:45/土日祝日及び当社休業日を除く)
製品情報問い合わせ先
グラクソ・スミスクライン株式会社
ヴィーブヘルスケア・カスタマー・サービス
東京都港区赤坂1-8-1
電話:0120-066-525 (9:00〜17:45/土日祝日及び当社休業日を除く)
26. 製造販売業者等
26.1 製造販売元
ヴィーブヘルスケア株式会社
東京都港区赤坂1-8-1
26.2 販売元
グラクソ・スミスクライン株式会社
東京都港区赤坂1-8-1