医薬品情報
| 総称名 | レベスティブ |
|---|---|
| 一般名 | テデュグルチド(遺伝子組換え) |
| 欧文一般名 | Teduglutide[Genetical Recombination] |
| 製剤名 | テデュグルチド(遺伝子組換え)皮下注用製剤 |
| 薬効分類名 | GLP-2アナログ製剤 |
| 薬効分類番号 | 2499 |
| ATCコード | A16AX08 |
| KEGG DRUG |
D06053
テデュグルチド
|
| JAPIC | 添付文書(PDF) |
添付文書情報2025年9月 改訂(第6版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| レベスティブ皮下注用3.8mg | Revestive 3.8mg for S.C.Injection | 武田薬品工業 | 2499419D1026 | 73683円/瓶 | 劇薬, 処方箋医薬品 |
| レベスティブ皮下注用0.95mg | Revestive 0.95mg for S.C.Injection | 武田薬品工業 | 2499419D2022 | 18421円/瓶 | 劇薬, 処方箋医薬品 |
2. 禁忌
次の患者には投与しないこと
4. 効能または効果
5. 効能または効果に関連する注意
5.1 本剤は腸管の順応期間を経て、経静脈栄養量及び補液量が安定した、あるいはそれ以上低減することが困難と判断された患者に投与すること。
5.2 修正月齢4ヵ月未満の患者を対象とした臨床試験は実施しておらず、投与は推奨されない。[9.7参照]
6. 用法及び用量
通常、テデュグルチド(遺伝子組換え)として1日1回0.05mg/kgを皮下注射する。
7. 用法及び用量に関連する注意
7.1 本剤の投与中は継続的に有効性を評価すること。成人では12ヵ月間の投与でも改善が認められない場合には、投与継続の必要性を検討すること。小児では投与6ヵ月後に有効性を評価し投与継続の必要性を検討すること。本剤投与中に経静脈栄養が不要になった患者においては、個々の患者の状況を踏まえて本剤の投与継続の必要性を検討すること。
7.2 中等度以上の腎機能障害(クレアチニンクリアランス50mL/min未満)患者では、本剤の血中濃度が上昇することから、1回あたりの投与量は0.025mg/kgとすること。[7.3、9.2.1、16.6.1参照]
7.3 下表を参照し患者の体重に応じて、投与製剤を選択すること。3.8mg製剤と0.95mg製剤との生物学的同等性試験は実施していないため、互換使用を行わないこと。[7.2、9.2.1、16.6.1参照]
| 患者体重 | 投与製剤(販売名) |
| ・体重10kg未満注) ・中等度以上の腎機能障害患者(クレアチニンクリアランス50mL/min未満)では体重20kg未満注) | レベスティブ皮下注用0.95mg |
| ・体重10kg以上20kg以下 ・中等度以上の腎機能障害患者(クレアチニンクリアランス50mL/min未満)では体重20kg以上40kg以下 | レベスティブ皮下注用0.95mg又は レベスティブ皮下注用3.8mg |
| ・体重20kg超 ・中等度以上の腎機能障害患者(クレアチニンクリアランス50mL/min未満)では体重40kg超 | レベスティブ皮下注用3.8mg |
7.4 投与を忘れた場合には、気づいた時点で直ちに投与すること。ただし、1日に2回の投与は行わないこと。
8. 重要な基本的注意
8.1 臨床試験において、大腸ポリープが報告されている。成人では、本剤の投与開始前の6ヵ月以内に大腸内視鏡検査又は他の画像検査等を実施し、大腸ポリープを認めた場合には投与開始前に切除を検討すること。投与開始後1年から2年の間に、大腸内視鏡検査又は他の画像検査等により経過を観察することが望ましい。大腸ポリープのリスクの高い患者では、必要に応じてその後も5年以内を目途に大腸内視鏡検査を行うこと。大腸ポリープを認めた場合には、最新のポリープの治療に関するガイドライン等を参考に適切な処置を行うこと。大腸癌と診断された場合には、投与を中止し適切な処置を行うこと。
1歳以上の小児では、本剤の投与開始前に便潜血検査を行うこと。原因不明の潜血が認められた場合には大腸内視鏡検査又は他の画像検査等を行い、大腸ポリープを認めた場合には投与開始前に切除を検討すること。便潜血検査で原因不明の潜血が認められた小児では、本剤投与中は年1回の頻度で便潜血検査を行うこと。1歳未満の小児では、実施可能性も考慮した上で投与開始前の便潜血検査及び大腸内視鏡検査等を実施すること。全ての小児で、本剤投与中は投与開始1年後、それ以降は5年ごと、及び原因不明の消化管出血が認められた場合には、大腸内視鏡検査又は他の画像検査等を行い、大腸ポリープの有無を確認することが望ましい。大腸ポリープ又は大腸癌を認めた場合は適切な処置を行うこと。
本剤投与終了後は必要に応じて便潜血検査及び大腸内視鏡検査等を実施すること。[2.2、2.3、11.1.1参照]
1歳以上の小児では、本剤の投与開始前に便潜血検査を行うこと。原因不明の潜血が認められた場合には大腸内視鏡検査又は他の画像検査等を行い、大腸ポリープを認めた場合には投与開始前に切除を検討すること。便潜血検査で原因不明の潜血が認められた小児では、本剤投与中は年1回の頻度で便潜血検査を行うこと。1歳未満の小児では、実施可能性も考慮した上で投与開始前の便潜血検査及び大腸内視鏡検査等を実施すること。全ての小児で、本剤投与中は投与開始1年後、それ以降は5年ごと、及び原因不明の消化管出血が認められた場合には、大腸内視鏡検査又は他の画像検査等を行い、大腸ポリープの有無を確認することが望ましい。大腸ポリープ又は大腸癌を認めた場合は適切な処置を行うこと。
本剤投与終了後は必要に応じて便潜血検査及び大腸内視鏡検査等を実施すること。[2.2、2.3、11.1.1参照]
8.2 本剤の薬理作用及び非臨床試験成績から、胃、小腸、肝胆道系及び膵臓にポリープや増殖性変化が認められる可能性があるので、本剤の投与開始前、投与中及び投与終了後は患者の状態を十分観察し、胃、小腸、肝胆道系又は膵臓に悪性腫瘍が認められた場合には、投与を中止し、適切な処置を行うこと。胃、小腸、肝胆道系又は膵臓に良性の腫瘍が認められた場合には、切除を検討する等、適切な処置を行うこと。[2.2、2.3、9.1.1参照]
8.3 胆嚢炎、胆管炎及び胆石症があらわれることがあるので、本剤投与開始前及び投与中は定期的に肝機能検査(ビリルビン、ALP等)や画像検査を行うこと。[11.1.3参照]
8.4 慢性膵炎、急性膵炎、膵管狭窄、膵感染等の膵疾患があらわれることがあるので、本剤投与開始前及び投与中は定期的に膵機能検査(リパーゼ、アミラーゼ等)や画像検査を行うこと。[11.1.4参照]
8.5 本剤投与により、消化管から吸収される水分量が増加し、体液量が過剰となり、うっ血性心不全があらわれることがある。一方で、短腸症候群の患者は脱水症になりやすいため、本剤投与中は経静脈栄養量を注意深く調整すること。特に本剤の投与開始から数ヵ月間、中止時、急激に電解質バランスや体液量が変動するおそれがある場合(脱水、感染、腸閉塞、術後等)には、電解質バランス及び体液量の状態を注意深く観察すること。また、急激な体重増加、顔面や下肢の浮腫、呼吸困難等が認められた場合には、医師に相談するよう患者又はその家族に指導すること。なお、臨床試験において、投与開始4週間後まで体液量の過剰が高い頻度で認められた。[11.1.5参照]
8.6 本剤の投与開始にあたっては、医療施設において、必ず医師によるか、医師の直接の監督のもとで投与を行うこと。また、以下の点に注意すること。
・自己投与の適用については、医師がその妥当性を慎重に検討し、十分な教育訓練を実施したのち、本剤投与による危険性と対処法について患者又はその家族が理解し、患者又はその家族が確実に投与できることを確認した上で、医師の管理指導のもとで実施すること。また、適用後、本剤による副作用が疑われる場合や自己投与の継続が困難な状況となる可能性がある場合には、直ちに自己投与を中止させ、医師の管理下で慎重に観察するなど適切な処置を行うこと。
・自己投与を適用する場合には、使用済みの注射針及び注射器を再使用しないように患者又はその家族に注意を促し、全ての器具の安全な廃棄方法に関する指導を行うと同時に、使用済みの注射針及び注射器を廃棄する容器を提供すること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 胃腸、肝胆道系及び膵臓以外に悪性腫瘍を有する患者
9.1.2 心不全及び高血圧等の心血管疾患の既往歴のある患者
9.2 腎機能障害患者
9.2.1 中等度以上の腎機能障害患者(クレアチニンクリアランス50mL/min未満)
9.5 妊婦
9.6 授乳婦
9.7 小児等
修正月齢4ヵ月未満の患者への投与は推奨されない。修正月齢4ヵ月未満の患者を対象とした臨床試験は実施していない。[5.2参照]
9.8 高齢者
10. 相互作用
相互作用序文
本剤の薬理作用により、併用する経口剤の吸収を高める可能性があるため、患者の状態を注意深く観察し、必要に応じて併用する経口剤の投与量を調整すること。特に、漸増投与が必要又は治療域が狭い経口剤を併用する場合には注意すること。
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 腸ポリープ(1.7%)
大腸ポリープ、十二指腸ポリープ等の腸ポリープがあらわれることがある。[8.1参照]
11.1.2 腸閉塞、消化管ストーマの閉塞(3.0%)
結腸狭窄、小腸狭窄等の腸閉塞、消化管ストーマの閉塞があらわれることがある。
11.1.3 胆嚢・胆道障害(1.7%)
胆嚢炎、急性胆嚢炎、胆管炎、胆石症等の胆嚢・胆道障害があらわれることがある。[8.3参照]
11.1.4 膵疾患(0.9%)
慢性膵炎、急性膵炎、膵管狭窄、膵感染等の膵疾患があらわれることがある。[8.4参照]
11.1.5 体液貯留(4.3%)
11.2 その他の副作用
| 10%以上 | 5〜10%未満 | 5%未満 | 頻度不明 | |
| 胃腸障害 | 腹痛 | 腹部膨満、悪心、嘔吐 | 鼓腸 | |
| 一般・全身障害および投与部位の状態 | 注射部位反応(注射部位紅斑、注射部位腫脹、注射部位疼痛等) | |||
| 免疫系障害 | 過敏症 | |||
| 感染症および寄生虫症 | インフルエンザ様疾患 | 鼻咽頭炎、インフルエンザ | ||
| 傷害、中毒および処置合併症 | 消化管ストーマ合併症(ストーマサイズの増大、ストーマ乳頭サイズの増大等) | |||
| 代謝および栄養障害 | 食欲減退 | |||
| 神経系障害 | 頭痛 | |||
| 精神障害 | 不眠症 | |||
| 呼吸器、胸郭および縦隔障害 | 咳嗽 |
14. 適用上の注意
14.1 薬剤交付時の注意
<3.8mg製剤>
14.1.1 患者が家庭で保存する場合においても、3.8mg製剤は凍結を避けて25℃以下で保存すること。
<0.95mg製剤>
14.1.2 患者が家庭で保存する場合においても、0.95mg製剤は凍結を避けて2〜8℃で冷蔵保存すること。やむを得ず0.95mg製剤を冷蔵保存できない場合には、凍結を避け25℃以下で使用期限を超えない範囲で6ヵ月以内に使用するよう指導すること。
14.2 薬剤調製時の注意
<3.8mg製剤>
<0.95mg製剤>
<製剤共通>
14.2.3 本剤に溶解液を加えた後、バイアルを掌に挟んで転がし、その後穏やかに1回反転させること。(激しく振とうしないこと)
14.2.4 調製後、濁り、微粒子、沈殿等が認められる場合には使用しないこと。
14.2.5 1バイアルあたり、注射液0.38mL又はそれ以上を注射用シリンジへ採取できる。注射液0.38mLはテデュグルチドの投与量として3.8mg製剤で3.8mg、0.95mg製剤で0.95mgに相当する。製剤間で注射液のテデュグルチド濃度が異なるため、用いる製剤を変更する際には注射液量に注意すること。[3.1、14.2.1、14.2.2、14.2.6参照]
14.2.6 注射液をバイアルから注射用シリンジに全量採取した後、3.8mg製剤はテデュグルチド10mg/mLの濃度で、0.95mg製剤はテデュグルチド2.5mg/mLの濃度で必要な注射液量へ調整すること。[3.1、14.2.1、14.2.2、14.2.5参照]
14.2.7 調製後は速やかに投与すること。本剤は保存剤を含有していないため、調製後は3時間以内に使用すること。未使用分は廃棄すること。
14.2.8 注射液は凍結させないこと。
14.3 薬剤投与時の注意
14.3.1 皮下注射は、腹部、大腿部又は上腕部に行うこと。投与部位は投与毎に変更すること。
14.3.2 皮膚が敏感な部位、皮膚に異常のある部位(傷、発疹、発赤、硬結等)には投与しないこと。
14.3.3 本剤は1回使用の製剤であり、再使用しないこと。
15. その他の注意
15.1 臨床使用に基づく情報
ペプチド製剤では免疫原性を示すことが知られており、テデュグルチド投与により抗体が発現する可能性がある。海外の臨床試験では1日1回テデュグルチド0.05mg/kgを皮下投与した被験者において、投与開始3ヵ月後で3%(2/60例)、6ヵ月後で17%(13/77例)、12ヵ月後で24%(16/67例)、24ヵ月後で33%(11/33例)、30ヵ月後で48%(14/29例)に抗テデュグルチド抗体の発現が確認された。国内の臨床試験では1日1回テデュグルチド0.05mg/kgを皮下投与した被験者において、投与開始6ヵ月後で1/6例、39ヵ月後で2/5例に抗テデュグルチド抗体の発現が確認された。抗テデュグルチド抗体の発現が確認された被験者において、臨床的に問題となる安全性の所見、効果の減弱又は薬物動態への影響は認められなかった7)。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
経静脈栄養を必要とする日本人成人短腸症候群患者7例を対象にテデュグルチド0.05mg/kgを単回皮下投与した時の血漿中テデュグルチド濃度推移及び薬物動態パラメータは以下のとおりであった8)。
単回投与時の血漿中濃度推移(平均値+標準偏差)
| Cmax(ng/mL) | 7例 | 49.5±16.43 |
| Tmax(h) | 7例 | 3.6±1.64 |
| AUC0-inf(ng・h/mL) | 6例 | 252±85.37 |
| t1/2(h) | 6例 | 1.1±0.21 |
| CL/F/weight(mL/h/kg) | 6例 | 229±101.4 |
| Vz/F/weight(mL/kg) | 6例 | 356±189.2 |
16.1.2 反復投与
(1)成人
経静脈栄養を必要とする成人短腸症候群患者12例を対象にテデュグルチド0.03〜0.15mg/kgを1日1回21日間皮下投与した時の薬物動態パラメータはDay 1及びDay 21で類似しており、蓄積性は認められなかった。また、0.03〜0.15mg/kgの範囲で用量比例性が認められた9)注)(外国人データ)。
注)本剤の承認用量は1日1回0.05mg/kgである。
| 0.03mg/kg(3例) | 0.10mg/kg(5例) | 0.15mg/kg(4例) | ||||
| Day 1 | Day 21 | Day 1 | Day 21 | Day 1 | Day 21 | |
| Cmax(ng/mL) | 39.7±16.9 | 43.4±10.5 | 122.5±64.6 | 111.9±30.5 | 179.4±119.9 | 138.5±70.9 |
| Tmax(h) | 3.3±2.3 | 2.7±0.3 | 3.1±0.7 | 2.7±0.6 | 3.3±0.6 | 3.5±0.7 |
| AUC0-inf(ng・h/mL) | 236.9注1) | − | 565.6±296.5 | − | 825.0±204.5 | − |
| AUCτ(ng・h/mL) | 139.7±86.1 | 148.5±42.8 | 539.0±286.7 | 533.0±197.9 | 792.3±217.4 | 698.8±261.7 |
| t1/2(h) | 2.0注1) | 1.2±0.9 | 1.6±0.9 | 1.2±0.1 | 2.0±0.7 | 1.5±0.1注2) |
(2)小児
修正月齢4ヵ月以上15歳以下の経静脈栄養を必要とする日本人短腸症候群患者を対象にテデュグルチド0.05mg/kgを反復皮下投与した時の血漿中テデュグルチド濃度は以下のとおりであった10)。
| 1歳以上(6例) | 1歳未満(2例) | ||
| Day 1 | 投与後1時間 | 23.7±12.8(6例) | 63.7注1)(2例) |
| 投与後6時間 | 6.1±4.6(4例) | 3.6注2)(1例) | |
| Week 4 | 投与後2時間 | 26.2±9.1(6例) | 31.2注1)(2例) |
| 投与後4時間 | 18.3±12.4(6例) | 7.2注1)(2例) | |
16.2 吸収
テデュグルチドを腹部に皮下投与後の絶対的バイオアベイラビリティは87.1%であった11)(外国人データ)。
16.3 分布
16.3.1 蛋白結合率
テデュグルチドのヒト血漿蛋白結合率は検討した濃度範囲(25、100、1000及び10000ng/mL)で、79.2%〜93.5%であった(in vitro)12)。
16.4 代謝
テデュグルチドは天然型GLP-2(グルカゴン様ペプチド-2)と同様に加水分解による代謝を受け、ペプチドやアミノ酸に分解されると考えられる。テデュグルチドはヒト肝細胞中で安定であり、肝細胞に関連する代謝経路はないことが示唆された(in vitro)13)。
16.5 排泄
健康被験者を対象にテデュグルチドを静脈内投与後のクリアランスは約123mL/h/kgで糸球体ろ過量(GFR)にほぼ等しく、テデュグルチドが主として腎臓を介して消失することが示唆された11)(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
中等度並びに重度腎機能障害及び末期腎不全の患者にテデュグルチド10mgを単回皮下投与した時の血漿中テデュグルチド濃度のAUC0-inf及びCmaxは、腎機能障害の重症度が高いほど高値となり、中等度腎機能障害及び末期腎不全の患者では腎機能正常者と比較してAUC0-infがそれぞれ1.5倍及び2.6倍、Cmaxがそれぞれ1.6倍及び2.1倍であった14)(外国人データ)。また、母集団薬物動態解析の結果、軽度腎機能障害の日本人患者でのCL/F及びVc/Fの平均値は腎機能正常の日本人患者での値との差が10%以内であったが、中等度腎機能障害の日本人患者では腎機能正常の日本人患者と比較してCL/F及びVc/Fがそれぞれ37%及び24%低かった15)。[7.2、7.3、9.2.1参照]
16.6.2 肝機能障害患者
中等度肝機能障害患者(Child-Pugh分類、グレードB)にテデュグルチド20mgを単回皮下投与した時の血漿中テデュグルチド濃度のCmax及びAUCは、中等度の肝障害を有する被験者の方が、肝機能正常者よりも10%〜15%低かった16)(外国人データ)。
16.6.3 高齢者
16.6.4 母集団薬物動態解析
外国人健康成人並びに日本人及び外国人の短腸症候群患者(成人及び小児)(478例)から得られた血漿中テデュグルチド濃度データ(6,775ポイント)を用いて母集団薬物動態解析を行った。日本人成人及び小児短腸症候群患者にテデュグルチド0.05mg/kgを1日1回反復皮下投与した時の母集団薬物動態解析の結果に基づき推定した薬物動態パラメータは以下のとおりであった17)18)。
| 18歳以上(14例) | 18歳未満(11例) | |
| Cmax,ss(ng/mL) | 46.3±11.0 | 32.2±11.9 |
| AUCss(ng・h/mL) | 232±52.9 | 102±31.1 |
| t1/2(h) | 1.10±0.304 | 0.890±0.294 |
| CL/F/weight(L/h/kg) | 0.228±0.0520 | 0.512±0.164 |
| Vc/F/weight(L/kg) | 0.348±0.0760 | 0.619±0.156 |
16.7 薬物相互作用
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 成人短腸症候群患者を対象とした第III相試験
(1)国内第III相試験(単群非盲検試験)
経静脈栄養を必要とする16歳以上の日本人短腸症候群患者7例を対象にテデュグルチド0.05mg/kgを1日1回24週間皮下投与した。なお、本試験では、最長8週間の経静脈サポート最適化期間及び4〜8週間の経静脈サポート安定化期間の後に、24週間の治験薬投与期間が設定された。また、被験者が自己投与方法を習得したと治験担当医師に判断された後に、自己投与を実施した。投与20週及び24週の両時点で週間経静脈栄養量のベースラインからの減少が20%以上を達成した被験者の割合は57.1%(4/7例)であった。24週における週間経静脈栄養量のベースラインからの変化量(平均値±標準偏差)及び変化率(平均値±標準偏差)は−3.3±3.61L/週及び−25.6±25.52%であった。本試験中に経静脈栄養からの完全離脱を達成した被験者は認められなかった。24週時点の週間経静脈栄養施行日数のベースラインからの変化日数(平均値±標準偏差)は−0.5±1.2日/週であった20)。
副作用発現頻度は、57.1%(4/7例)であり、主な副作用は、腹部膨満28.6%(2/7例)であった21)。
副作用発現頻度は、57.1%(4/7例)であり、主な副作用は、腹部膨満28.6%(2/7例)であった21)。
(2)海外第III相試験(二重盲検比較試験)
経静脈栄養を必要とする18歳以上の短腸症候群患者86例を対象に、テデュグルチド0.05mg/kgを1日1回もしくはプラセボを皮下投与した。なお、本試験では、最長8週間の経静脈サポート最適化期間、4〜8週間の経静脈サポート安定化期間及び24週間の治験薬投与期間が設定された。また、被験者が自己投与方法を習得したと治験担当医師に判断された後に、自己投与を実施した。投与20週及び24週の両時点で週間経静脈栄養量のベースラインからの減少が20%以上を達成した被験者の割合はテデュグルチド0.05mg/kg群で62.8%(27/43例)、プラセボ群で30.2%(13/43例)であり、テデュグルチド0.05mg/kg群のプラセボ群に対する統計学的有意差が示された(p=0.002、Cochran-Mantel-Haenszel検定)22)。24週における週間経静脈栄養量のベースラインからの変化量(平均値±標準偏差)及び変化率(平均値±標準偏差)は、テデュグルチド0.05mg/kg群で−4.4±3.8L/週及び−32.4±18.9%、プラセボ群で−2.3±2.7L/週及び−21.3±25.4%であった23)。本試験中に経静脈栄養からの完全離脱を達成した被験者は認められなかった。24週において週間経静脈栄養施行日数にベースラインから1日以上の減少がみられた被験者の割合は、テデュグルチド0.05mg/kg群で53.8%(21/39例)、プラセボ群で23.1%(9/39例)であった22)。
副作用発現頻度は、54.8%(23/42例)であった。主な副作用は、消化管ストーマ合併症23.8%(10/42例)、腹部膨満、腹痛、悪心がそれぞれ16.7%(7/42例)であった22)。
副作用発現頻度は、54.8%(23/42例)であった。主な副作用は、消化管ストーマ合併症23.8%(10/42例)、腹部膨満、腹痛、悪心がそれぞれ16.7%(7/42例)であった22)。
17.1.2 小児短腸症候群患者を対象とした第III相試験
(1)国内第III相試験(単群非盲検試験)
経静脈栄養を必要とする修正月齢4ヵ月〜15歳までの日本人小児短腸症候群患者10例〔小児8例(1〜15歳)及び乳児2例(4〜12ヵ月未満)〕を対象にテデュグルチド0.05mg/kgを1日1回24週間皮下投与した。なお、被験者が自己投与方法を習得したと治験担当医師に判断された後に、自己投与を実施した。解析対象とした8例(小児6例、乳児2例)について、週間経静脈栄養量のベースラインからの減少が20%以上を達成した被験者の割合は小児被験者で66.7%(4/6例)、乳児被験者で50.0%(1/2例)であった。投与終了時における経静脈栄養量のベースラインからの変化量(平均値±標準偏差)及び変化率(平均値±標準偏差)は、小児被験者において−14.5±8.03mL/kg/日及び−35.7±33.18%、乳児被験者において−26.2±13.61mL/kg/日及び−26.7±15.14%であった。小児被験者1例にて、経静脈栄養からの完全離脱を達成した。乳児被験者において経静脈栄養からの完全離脱を達成した被験者は認められなかった24)。小児被験者における投与終了時の週間経静脈栄養施行日数のベースラインからの変化日数(平均値±標準偏差)は−1.2±2.86日/週であった25)。乳児被験者では経静脈栄養施行日数の減少はみられなかった24)。
副作用発現頻度は、小児で75.0%(6/8例)、乳児で0%(0/2例)であった。主な副作用(小児)は、注射部位紅斑37.5%(3/8例)、腹痛25.0%(2/8例)であった24)。
副作用発現頻度は、小児で75.0%(6/8例)、乳児で0%(0/2例)であった。主な副作用(小児)は、注射部位紅斑37.5%(3/8例)、腹痛25.0%(2/8例)であった24)。
(2)海外第III相試験(二重盲検比較試験)
経静脈栄養を必要とする1〜17歳までの短腸症候群患者59例を対象にテデュグルチド0.025mg/kg注)又は0.05mg/kgを1日1回皮下投与もしくは標準的な薬物療法を24週間行った。なお、被験者が自己投与方法を習得したと治験担当医師に判断された後に、自己投与を実施した。投与終了時において週間経静脈栄養量のベースラインからの減少が20%以上を達成した被験者の割合は、テデュグルチド0.05mg/kg/日群で69.2%(18/26例)、標準治療群で11.1%(1/9例)であった。投与終了時における経静脈栄養量のベースラインからの変化量(平均値±標準偏差)及び変化率(平均値±標準偏差)は、テデュグルチド0.05mg/kg/日群で−23.30±17.50mL/kg/日及び−41.57±28.90%、標準治療群で−6.03±4.55mL/kg/日及び−10.21±13.59%であった。投与終了時までにテデュグルチド0.05mg/kg/日群の11.5%(3/26例)が経静脈栄養からの完全離脱を達成した。標準治療群では経静脈栄養からの完全離脱を達成した被験者は認められなかった。テデュグルチド0.05mg/kg/日群における投与終了時の週間経静脈栄養施行日数のベースラインからの変化日数(平均値±標準偏差)は−1.34±2.24日/週であった。標準治療群では、週間経静脈栄養施行日数は減少しなかった26)。
副作用発現頻度は、テデュグルチド0.05mg/kg群で26.9%(7/26例)であった26)。
副作用発現頻度は、テデュグルチド0.05mg/kg群で26.9%(7/26例)であった26)。
注)本剤の承認用量は1日1回0.05mg/kgである。
18. 薬効薬理
18.1 作用機序
18.2 In vitro活性
18.3 腸管に対する作用
19. 有効成分に関する理化学的知見
19.1. テデュグルチド(遺伝子組換え)
| 一般的名称 | テデュグルチド(遺伝子組換え) |
|---|---|
| 一般的名称(欧名) | Teduglutide[Genetical Recombination] |
| 分子式 | C164H252N44O55S |
| 分子量 | 3,752.08 |
| 理化学知見その他 | テデュグルチドは、遺伝子組換えヒトグルカゴン様ペプチド-2(GLP-2)類縁体であり、2番目のAlaがGlyに置換されている。テデュグルチドは、33個のアミノ酸残基からなるペプチドである。 |
| KEGG DRUG |
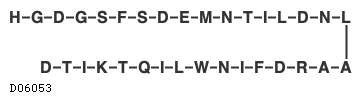
|
21. 承認条件
21.1 医薬品リスク管理計画を策定の上、適切に実施すること。
21.2 国内での治験症例が極めて限られていることから、製造販売後、一定数の症例に係るデータが集積されるまでの間は、全症例を対象に使用成績調査を実施することにより、本剤使用患者の背景情報を把握するとともに、本剤の安全性及び有効性に関するデータを早期に収集し、本剤の適正使用に必要な措置を講じること。
22. 包装
<レベスティブ皮下注用3.8mg>
1バイアル(日局注射用水0.5mL入りシリンジ1本及びバイアルアダプター1個添付)
<レベスティブ皮下注用0.95mg>
1バイアル(日局注射用水0.5mL入りシリンジ1本及びバイアルアダプター1個添付)
23. 主要文献
- テデュグルチドの反復投与毒性試験(2021年6月23日承認、CTD2.6.6.3)
- テデュグルチドのがん原性試験(2021年6月23日承認、CTD2.6.6.5)
- テデュグルチドの有害事象(2021年6月23日承認、CTD2.7.4.2.1.5.2)
- テデュグルチドのラット胚・胎児発生毒性試験(2021年6月23日承認、CTD2.6.6.6.2.1)
- テデュグルチドの胎盤通過及び乳汁移行(2021年6月23日承認、CTD2.6.4.4.4)
- テデュグルチドのラット出生前/出生後の発生並びに母体機能に関する試験(2021年6月23日承認、CTD2.6.6.6.3.1)
- テデュグルチドの免疫原性(2021年6月23日承認、CTD2.7.2.4.1)
- テデュグルチドの薬物動態試験成績[1](2021年6月23日承認、CTD2.7.2.2.2.3.1.2.2)
- テデュグルチドの薬物動態試験成績[2](2021年6月23日承認、CTD2.7.2.2.2.3.1.1.1)
- テデュグルチドの薬物動態試験成績[3](2021年6月23日承認、CTD2.7.2.2.2.3.2.2.1)
- テデュグルチドの薬物動態試験成績[4](2021年6月23日承認、CTD2.7.1.2.1.1)
- テデュグルチドの血漿蛋白結合率(2021年6月23日承認、CTD2.6.4.4.3)
- テデュグルチドの代謝安定性(2021年6月23日承認、CTD2.6.4.5.1.1)
- テデュグルチドの薬物動態試験成績[5](2021年6月23日承認、CTD2.7.2.2.2.2.2)
- テデュグルチドの母集団薬物動態解析[1](2021年6月23日承認、CTD2.7.2.3.3.2)
- テデュグルチドの薬物動態試験成績[6](2021年6月23日承認、CTD2.7.2.2.2.2.1)
- テデュグルチドの母集団薬物動態解析[2](2021年6月23日承認、CTD2.7.2.3.2)
- テデュグルチドの母集団薬物動態解析[3](2021年6月23日承認、CTD2.7.2.3.3.4)
- テデュグルチドの薬物動態学的薬物相互作用(2021年6月23日承認、CTD2.6.4.7)
- テデュグルチドの国内第III相臨床試験成績[1](2021年6月23日承認、CTD2.7.3.2)
- テデュグルチドの国内第III相臨床試験成績[2](2021年6月23日承認、CTD2.7.6.16)
- テデュグルチドの海外第III相臨床試験成績[1](2021年6月23日承認、CTD2.7.6.13)
- テデュグルチドの海外第III相臨床試験成績[2](2021年6月23日承認、CTD2.7.3.3)
- テデュグルチドの国内第III相臨床試験成績[3](2021年6月23日承認、CTD2.7.6.23)
- テデュグルチドの国内第III相臨床試験成績[4](2021年6月23日承認、CTD2.7.3.3)
- テデュグルチドの海外第III相臨床試験成績[3](2021年6月23日承認、CTD2.7.6.18)
- Cheeseman CI,et al., Am J Physiol., 271, G477-G482, (1996) »PubMed
- Cottrell JJ,et al., Am J Physiol Gastrointest Liver Physiol., 290, G293-G300, (2006) »PubMed
- Dube PE,et al., Am J Physiol Endocrinol Metab., 293, E460-E465, (2007) »PubMed
- Orskov C,et al., Regul Pept., 124, 105-112, (2005) »PubMed
- Yusta B,et al., J Biol Chem., 274, 30459-30467, (1999) »PubMed
- Demchyshyn LL,et al., Gastroenterology., 116 (suppl.2), A545, (1999)
- Demchyshyn LL,et al., Gastroenterology., 120 (suppl.1), A509, (2001)
- テデュグルチドの腸管栄養活性、腸管粘膜バリア及び吸収に対する作用(2021年6月23日承認、CTD2.6.2.2.4、2.6.2.2.5)
- テデュグルチドのTPN誘発性腸形成不全モデルに対する効果(2021年6月23日承認、CTD2.6.2.2.6)
24. 文献請求先及び問い合わせ先
文献請求先
武田薬品工業株式会社
くすり相談室
〒103-8668
東京都中央区日本橋本町二丁目1番1号
電話:フリーダイヤル 0120-566-587 受付時間 9:00〜17:30(土日祝日・弊社休業日を除く)
製品情報問い合わせ先
武田薬品工業株式会社
くすり相談室
〒103-8668
東京都中央区日本橋本町二丁目1番1号
電話:フリーダイヤル 0120-566-587 受付時間 9:00〜17:30(土日祝日・弊社休業日を除く)
26. 製造販売業者等
26.1 製造販売元
武田薬品工業株式会社
〒540-8645
大阪市中央区道修町四丁目1番1号