医薬品情報
| 総称名 | ダパグリフロジン |
|---|---|
| 一般名 | ダパグリフロジン |
| 欧文一般名 | Dapagliflozin |
| 製剤名 | ダパグリフロジン錠 |
| 薬効分類名 | 選択的SGLT2阻害剤 2型糖尿病治療剤 |
| 薬効分類番号 | 3969 |
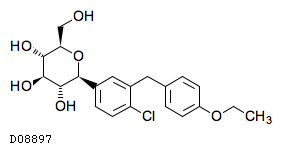
| KEGG DRUG |
D08897
ダパグリフロジン
|
| KEGG DGROUP |
DG00122
ダパグリフロジン
DG01794
SGLT2阻害薬
DG02044
血糖降下薬
|
| JAPIC | 添付文書(PDF) |
添付文書情報2026年2月 作成(第1版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ダパグリフロジン錠5mg「EP」 | DAPAGLIFLOZIN TABLETS「EP」 | 第一三共エスファ | 39690B6F1051 | 処方箋医薬品注) | |
| ダパグリフロジン錠10mg「EP」 | DAPAGLIFLOZIN TABLETS「EP」 | 第一三共エスファ | 39690B6F2058 | 処方箋医薬品注) |
2. 禁忌
次の患者には投与しないこと
2.1 本剤の成分に対し過敏症の既往歴のある患者
2.2 重症ケトーシス、糖尿病性昏睡又は前昏睡の患者[輸液、インスリンによる速やかな高血糖の是正が必須となるので本剤の投与は適さない。]
2.3 重症感染症、手術前後、重篤な外傷のある患者[インスリン注射による血糖管理が望まれるので本剤の投与は適さない。]
4. 効能または効果
5. 効能または効果に関連する注意
5.1 本剤は2型糖尿病と診断された患者に対してのみ使用し、1型糖尿病の患者には投与しないこと。
5.4 本剤の適用はあらかじめ糖尿病治療の基本である食事療法、運動療法を十分に行った上で効果が不十分な場合に限り考慮すること。
6. 用法及び用量
通常、成人にはダパグリフロジンとして5mgを1日1回経口投与する。なお、効果不十分な場合には、経過を十分に観察しながら10mg1日1回に増量することができる。
8. 重要な基本的注意
8.2 本剤投与中に、血清クレアチニンの上昇又はeGFRの低下がみられることがあるので、腎機能を定期的に検査すること。腎機能障害のある患者では経過を十分に観察し、継続的にeGFRが45mL/min/1.73m2未満に低下した場合は投与の中止を検討すること。[5.2、5.3、9.2.1、9.2.2、17.1.1参照]
8.3 本剤の利尿作用により多尿・頻尿がみられることがある。また、体液量が減少することがあるので観察を十分に行い、適度な水分補給を行うよう指導すること。特に体液量減少を起こしやすい患者(高齢者、腎機能障害のある患者、利尿剤併用患者等)においては、脱水や糖尿病ケトアシドーシス、高浸透圧高血糖症候群、脳梗塞を含む血栓・塞栓症等の発現に注意すること。[9.1.1、9.2.2、9.8、10.2、11.1.3、11.1.4参照]
8.4 本剤投与中は、血糖を定期的に検査するとともに、経過を十分に観察し、常に投与継続の必要性について注意を払うこと。本剤を3ヵ月投与しても効果が不十分な場合、より適切と考えられる治療を考慮すること。
8.5 尿路感染及び性器感染を起こし、腎盂腎炎、外陰部及び会陰部の壊死性筋膜炎(フルニエ壊疽)、敗血症等の重篤な感染症に至ることがある。尿路感染及び性器感染の症状及びその対処方法について患者に説明すること。[9.1.2、11.1.2参照]
8.6 本剤の作用機序である尿中グルコース排泄促進作用により、血糖コントロールが良好であっても脂肪酸代謝が亢進し、ケトーシスがあらわれ、ケトアシドーシスに至ることがある。
8.6.1 著しい血糖の上昇を伴わない場合があるため、以下の点に留意すること。[11.1.4参照]
(1)悪心・嘔吐、食欲減退、腹痛、過度な口渇、倦怠感、呼吸困難、意識障害等の症状が認められた場合には、血中又は尿中ケトン体測定を含む検査を実施すること。異常が認められた場合には投与を中止し、適切な処置を行うこと。
(2)特に、インスリン分泌能の低下、インスリン製剤の減量や中止、過度な糖質摂取制限、食事摂取不良、感染症、脱水を伴う場合にはケトアシドーシスを発現しやすいので、観察を十分に行うこと。
(3)患者に対し、以下の点を指導すること。
・ケトアシドーシスの症状(悪心・嘔吐、食欲減退、腹痛、過度な口渇、倦怠感、呼吸困難、意識障害等)。
・ケトアシドーシスの症状が認められた場合には直ちに医療機関を受診すること。
・血糖値が高値でなくともケトアシドーシスが発現しうること。
8.6.2 本剤を含むSGLT2阻害薬の投与中止後、血漿中半減期から予想されるより長く尿中グルコース排泄及びケトアシドーシスが持続した症例が報告されているため、必要に応じて尿糖を測定するなど観察を十分に行うこと。[11.1.4参照]
8.7 排尿困難、無尿、乏尿あるいは尿閉の症状を呈する患者においては、それらの治療を優先するとともに他剤での治療を考慮すること。
8.8 本剤投与による体重減少が報告されているため、過度の体重減少に注意すること。
8.9 低血糖症状を起こすことがあるので、高所作業、自動車の運転等に従事している患者に投与するときは注意すること。[11.1.1参照]
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 脱水を起こしやすい患者(血糖コントロールが極めて不良の患者、高齢者、利尿剤併用患者等)
9.1.2 尿路感染、性器感染のある患者
9.2 腎機能障害患者
9.2.1 重度の腎機能障害患者又は透析中の末期腎不全患者
9.2.2 中等度の腎機能障害患者
9.3 肝機能障害患者
重度の肝機能障害のある患者を対象とした臨床試験は実施していない。
9.5 妊婦
妊婦又は妊娠している可能性のある女性には本剤を投与せず、インスリン製剤等を使用すること。妊娠中の投与に関する安全性は確立されていない。動物実験(ラット)において、ヒトの妊娠中期及び後期にあたる期間の曝露及び生後21日〜90日の曝露により、出生児及び幼若動物に腎盂及び尿細管の拡張が認められたとの報告がある。また、本薬の動物実験(ラット)で胎児への移行が報告されている。
9.6 授乳婦
授乳しないことが望ましい。ラットで乳汁中への移行が報告されている。
9.7 小児等
小児等を対象とした有効性及び安全性を指標とした臨床試験は実施していない。
9.8 高齢者
10. 相互作用
相互作用序文
本剤は主として、UGT1A9によるグルクロン酸抱合により代謝される。[16.4参照]
薬物代謝酵素用語
UGT1A9
10.2 併用注意
| 糖尿病用薬 インスリン製剤 スルホニルウレア剤 チアゾリジン系薬剤 ビグアナイド系薬剤 α-グルコシダーゼ阻害剤 速効型インスリン分泌促進剤 DPP-4阻害剤 GLP-1受容体作動薬 等 [11.1.1参照] | 低血糖の発現に注意すること。特に、インスリン製剤、スルホニルウレア剤又は速効型インスリン分泌促進剤の減量を検討すること。 | 血糖降下作用が相加的に増強するおそれがある。 |
| 血糖降下作用を増強する薬剤 β遮断薬 サリチル酸剤 モノアミン酸化酵素阻害剤 等 | 併用時は血糖コントロールに注意し、血糖値、その他患者の状態を十分に観察しながら投与すること。 | 血糖降下作用が増強される。 |
| 血糖降下作用を減弱する薬剤 副腎皮質ホルモン 甲状腺ホルモン アドレナリン 等 | 併用時は血糖コントロールに注意し、血糖値、その他患者の状態を十分に観察しながら投与すること。 | 血糖降下作用が減弱される。 |
| 利尿薬 ループ利尿薬 サイアザイド系利尿薬 等 [8.3、9.1.1、11.1.3、16.7.2参照] | 必要に応じ利尿薬の用量を調整するなど注意すること。 | 利尿作用が増強される。 |
| リチウム製剤 炭酸リチウム | リチウムの作用が減弱されるおそれがある。 | リチウムの腎排泄を促進することにより、血清リチウム濃度が低下する可能性がある。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 低血糖(頻度不明)
11.1.2 腎盂腎炎(0.1%未満)注1)、外陰部及び会陰部の壊死性筋膜炎(フルニエ壊疽)(頻度不明)注1)、敗血症(0.1%未満)注1)
11.1.3 脱水(頻度不明)注1)
11.1.4 ケトアシドーシス(頻度不明)
2型糖尿病患者を対象とした臨床試験(D1692C00005試験、D1692C00006試験及びD1692C00012試験)等の合算により算出した。
注1)2型糖尿病患者を対象とした臨床試験(D1692C00005試験、D1692C00006試験及びD1692C00012試験)等の重篤な副作用の合算により算出した。
11.2 その他の副作用
| 5%以上 | 1〜5%未満 | 1%未満 | 頻度不明 | |
| 感染症 | 性器感染(腟カンジダ症等) | 尿路感染(膀胱炎等) | ||
| 血液 | ヘマトクリット増加 | |||
| 代謝及び栄養障害 | 体液量減少注2) | ケトーシス、食欲減退、多飲症 | ||
| 消化器 | 便秘、口渇 | 下痢、腹痛、悪心、嘔吐 | ||
| 筋・骨格系 | 背部痛、筋痙縮 | |||
| 皮膚 | 発疹 | |||
| 腎臓 | 頻尿、尿量増加 | 腎機能障害、排尿困難 | ||
| 精神神経系 | 頭痛、振戦、めまい | |||
| 眼 | 眼乾燥 | |||
| 生殖器 | 陰部そう痒症 | 外陰腟不快感 | ||
| 循環器 | 高血圧、低血圧 | |||
| その他 | 倦怠感、無力症、体重減少、異常感 |
12. 臨床検査結果に及ぼす影響
本剤の作用機序により、本剤服用中は尿糖陽性、血清1,5-AG(1,5-アンヒドログルシトール)低値を示す。尿糖及び血清1,5-AGの検査結果は、血糖コントロールの参考とはならないので注意すること。
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
15. その他の注意
15.1 臨床使用に基づく情報
2型糖尿病患者における国内外の臨床試験の併合解析において、全ての悪性腫瘍の発現割合は本剤群と対照群で同様であったが、膀胱癌及び乳癌では本剤群で多い傾向が認められた。しかしながら、投与開始から膀胱癌及び乳癌の診断までが短期間であったことから、いずれの腫瘍においても本剤との因果関係は確立されていない。
15.2 非臨床試験に基づく情報
発癌性あるいは変異原性は認められていない。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
16.1.2 反復投与
2型糖尿病患者9例にダパグリフロジン錠2.5注)及び10mgを1日1回14日間反復経口投与したとき、投与14日目における投与後のCmaxは48及び191ng/mL、AUCτは157及び727ng・hr/mLであり、累積係数は1.28及び1.21であった1)。
16.1.3 食事の影響
健康成人29例にダパグリフロジン錠10mgを空腹時又は高脂肪高カロリー食摂取後(食後)に投与したとき、空腹時投与に対する食後投与のダパグリフロジンのCmax及びAUCinfの幾何平均比(90%信頼区間)は、それぞれ0.550(0.499,0.606)及び0.973(0.943,1.004)であった。食後投与のTmaxの中央値は、空腹時投与と比べ1.25時間遅延した(外国人データ)3)。
16.1.4 生物学的同等性試験
<ダパグリフロジン錠10mg「EP」>
ダパグリフロジン錠10mg「EP」とフォシーガ錠10mgを、クロスオーバー法によりそれぞれ1錠(ダパグリフロジンとして10mg)健康成人男子に絶食時単回経口投与して血漿中未変化体濃度を測定し、得られた薬物動態パラメータ(AUC、Cmax)について90%信頼区間法にて統計解析を行った結果、log(0.80)〜log(1.25)の範囲内であり、両製剤の生物学的同等性が確認された4)。
血漿中濃度の推移
| AUC0-48(ng・hr/mL) | Cmax(ng/mL) | Tmax(hr) | t1/2(hr) | |
| ダパグリフロジン錠10mg「EP」 | 577.27±136.74 | 188.63±49.04 | 1.0±0.5 | 9.80±5.78 |
| フォシーガ錠10mg | 583.60±136.06 | 194.38±52.28 | 0.9±0.4 | 9.87±3.51 |
血漿中濃度並びにAUC、Cmax等のパラメータは、被験者の選択、血液の採取回数・時間等の試験条件によって異なる可能性がある。
16.2 吸収
健康成人男性7例にダパグリフロジン錠10mgを空腹時に経口投与し、その1時間後に[14C]ダパグリフロジン80μgを1分間かけて静脈内投与したとき、バイオアベイラビリティは78%であった(外国人データ)5)。
16.3 分布
16.4 代謝
16.5 排泄
外国人健康成人男性に50mgの[14C]ダパグリフロジンを投与したとき、総放射能の75%が尿中に、21%が糞中に排泄された。糞中からは投与量の約15%が未変化体として排泄された(外国人データ)11)13)。健康成人男性6例にダパグリフロジン2.5注)及び10mgを空腹時に単回経口投与したとき、未変化体として投与量の1.0%及び1.1%が投与120時間後までに尿中排泄された1)。2型糖尿病患者9例にダパグリフロジン錠2.5注)及び10mgを1日1回14日間反復投与したとき、未変化体として投与量の1.7%及び1.9%が投与24時間後までに尿中排泄された1)。
In vitroにおいて、ダパグリフロジンは有機アニオントランスポーター(OAT3)及び有機アニオントランスポーターポリペプチド(OATP1B1及びOATP1B3)に対して弱い阻害作用を示した(IC50値はそれぞれ33μM、69μM、8μM)。ダパグリフロジンはP-糖蛋白の弱い基質となるが、P-糖蛋白を阻害しなかった12)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害
健康成人及び2型糖尿病患者にダパグリフロジン錠50mg注)を単回投与したとき、腎機能が正常な被験者(健康成人(8例)及びCLcr>80mL/minである2型糖尿病患者(12例))に対する、軽度腎機能障害患者(50<CLcr≦80mL/minである2型糖尿病患者(8例))、中等度腎機能障害患者(30≦CLcr≦50mL/minである2型糖尿病患者(8例))及び重度腎機能障害患者(CLcr<30mL/minであり透析を受けていない2型糖尿病患者(4例))のCmax及びAUCinfの幾何平均の比(90%信頼区間)は、それぞれ1.142(1.052,1.239)及び1.278(1.189,1.374)、1.256(1.091,1.445)及び1.523(1.346,1.724)並びに1.355(1.123,1.633)及び1.753(1.486,2.068)であった(外国人データ)7)14)。
16.6.2 肝機能障害
健康成人及び肝機能障害者にダパグリフロジン錠10mgを単回投与したとき、健康成人(6例)に対する軽度(Child-Pugh分類でA(6例))、中等度(Child-Pugh分類でB(6例))及び重度(Child-Pugh分類でC(6例))の肝機能障害者におけるダパグリフロジンのCmax及びAUCinfの幾何平均の比(90%信頼区間)は、それぞれ0.882(0.598,1.301)及び1.033(0.765,1.396)、1.122(0.76,1.654)及び1.359(1.007,1.836)並びに1.395(0.946,2.056)及び1.669(1.236,2.255)であった(外国人データ)8)15)。
16.7 薬物相互作用
16.7.1 糖尿病用薬
16.7.2 利尿薬
16.7.3 その他の薬剤
16.8 その他
<ダパグリフロジン錠5mg「EP」>
ダパグリフロジン錠5mg「EP」は、「含量が異なる経口固形製剤の生物学的同等性試験ガイドライン(令和2年3月19日 薬生薬審発0319第1号)」に基づき、ダパグリフロジン錠10mg「EP」を標準製剤としたとき、溶出挙動に基づき生物学的に同等とみなされた24)。
注)本剤の承認用量は5〜10mg/日である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 2型糖尿病患者を対象とした試験
(1)国内臨床試験
国内の臨床試験において、ダパグリフロジン錠5mg又は10mgを1日1回投与した1012例中172例(17.0%)に副作用が認められた。主な副作用は、頻尿36例(3.6%)、口渇18例(1.8%)、性器感染17例(1.7%)、尿路感染17例(1.7%)等であった25)。(初回承認時)
・用量反応試験(単独療法、D1692C00005試験)
・プラセボ対照二重盲検比較試験(単独療法、D1692C00006試験)
ダパグリフロジン錠5及び10mgの24週間投与によりHbA1c変化量の結果は以下のとおりであり、プラセボに比べて有意に低下した。また、体重のプラセボとの差〔平均値±標準誤差〕は、5及び10mg群でそれぞれ−1.29±0.35kg及び−1.38±0.35kgであった。低血糖の有害事象発現割合は、プラセボ群で0%(0例/87例)、5mg群で0%(0例/86例)、10mg群で2.3%(2例/88例)であり、重度の低血糖は認められなかった28)29)30)。
| HbA1c(NGSP値)(%) | 空腹時血糖(mg/dL) | ||||
| ベースライン平均値(SD) | ベースライン値からの変化量注1)(SE) | プラセボとの差(SE) | ベースライン値からの変化量注1)(SE) | プラセボとの差(SE) | |
| プラセボ(n=87) | 7.50(0.63) | −0.06(0.06) | − | 5.8(2.17) | − |
| ダパグリフロジン錠5mg(n=86) | 7.50(0.72) | −0.41(0.06) | −0.35※(0.09) | −8.6(2.19) | −14.4※(2.90) |
| ダパグリフロジン錠10mg(n=88) | 7.46(0.61) | −0.45(0.06) | −0.39※(0.09) | −13.7(2.15) | −19.5※(2.89) |
腎機能別のHbA1c変化量の結果は以下のとおりであった30)31)。
| HbA1c(NGSP値)(%) | |||
| ベースライン平均値(SD) | ベースライン値からの変化量注2)[両側95%信頼区間] | プラセボとの差(SE) | |
| eGFR 60以上90mL/min/1.73m2未満 | |||
| プラセボ(n=57) | 7.59(0.63) | −0.01[−0.15,0.14] | − |
| ダパグリフロジン錠5mg(n=61) | 7.52(0.79) | −0.37[−0.51,−0.23] | −0.37(0.10) |
| ダパグリフロジン錠10mg(n=61) | 7.43(0.58) | −0.50[−0.64,−0.36] | −0.49(0.10) |
| eGFR 45以上60mL/min/1.73m2未満 | |||
| プラセボ(n=24) | 7.34(0.62) | −0.10[−0.32,0.13] | − |
| ダパグリフロジン錠5mg(n=23) | 7.44(0.53) | −0.46[−0.69,−0.23] | −0.37(0.16) |
| ダパグリフロジン錠10mg(n=24) | 7.55(0.70) | −0.31[−0.53,−0.08] | −0.21(0.16) |
・非盲検長期投与試験(単独及び他の糖尿病用薬との併用療法、D1692C00012試験)
ダパグリフロジン錠5mg(10mgへの増量を含む)の単独及び併用療法によるHbA1c変化量の結果は以下のとおりであった。また、単独療法群における52週時のベースライン値からの空腹時血糖変化量〔平均値±標準偏差〕は、−14.3±21.4mg/dL、体重変化量〔平均値±標準偏差〕は、−2.58±2.29kgであった。低血糖の有害事象発現割合は、単独療法群2.4%(6例/249例)、スルホニルウレア剤併用群6.6%(8例/122例)、DPP-4阻害剤併用群3.2%(2例/62例)、α-グルコシダーゼ阻害剤併用群0%(0例/61例)、ビグアナイド系薬剤併用群2.8%(2例/71例)、チアゾリジン系薬剤併用群1.6%(1例/64例)、速効型インスリン分泌促進剤併用群6.1%(3例/49例)、GLP-1受容体作動薬併用群6.0%(3例/50例)であった。重度の低血糖は認められなかった32)33)。
| HbA1c(NGSP値)(%) | ||
| ベースライン平均値(SD) | ベースライン値からの変化量(SD) | |
| ダパグリフロジン錠単独療法群(n=249) | 7.53(0.76) | −0.66(0.71) |
| スルホニルウレア剤併用群(n=122) | 8.02(0.84) | −0.65(0.70) |
| DPP-4阻害剤併用群(n=62) | 7.80(0.91) | −0.60(0.57) |
| α-グルコシダーゼ阻害剤併用群(n=61) | 7.59(0.73) | −0.81(0.67) |
| ビグアナイド系薬剤併用群(n=69) | 7.63(0.85) | −0.63(0.69) |
| チアゾリジン系薬剤併用群(n=64) | 7.94(0.92) | −0.86(0.76) |
| 速効型インスリン分泌促進剤併用群(n=49) | 7.49(0.73) | −0.76(0.65) |
| GLP-1受容体作動薬併用群(n=50) | 8.11(0.92) | −0.49(0.80) |
ダパグリフロジン錠単独療法群の腎機能別のHbA1c変化量の結果は以下のとおりであった30)。
| HbA1c(NGSP値)(%) | |
| ベースライン値からの変化量(SD) | |
| eGFR 90mL/min/1.73m2以上 | |
| ダパグリフロジン錠単独療法群(n=13) | −0.86(0.78) |
| eGFR 60以上90mL/min/1.73m2未満 | |
| ダパグリフロジン錠単独療法群(n=175) | −0.73(0.63) |
| eGFR 45以上60mL/min/1.73m2未満 | |
| ダパグリフロジン錠単独療法群(n=61) | −0.43(0.85) |
(2)海外臨床試験
・外国人の中等度腎機能障害患者を対象としたプラセボ対照二重盲検試験(単独療法、MB102029試験)
外国人の中等度腎機能障害患者(eGFRが30以上60mL/min/1.73m2未満)におけるHbA1c変化量の結果は以下のとおりであった。
| HbA1c(NGSP値)(%) | |||
| ベースライン平均値(SD) | ベースライン値からの変化量注3)[両側95%信頼区間] | プラセボとの差(SE) | |
| 全体 | |||
| プラセボ(n=82) | 8.53(1.29) | −0.32[−0.66,−0.01] | − |
| ダパグリフロジン錠5mg(n=83) | 8.30(1.04) | −0.41[−0.74,−0.07] | −0.08(0.14) |
| ダパグリフロジン錠10mg(n=82) | 8.22(0.97) | −0.44[−0.77,−0.10] | −0.11(0.15) |
| eGFR30以上45mL/min/1.73m2未満 | |||
| プラセボ(n=33) | 8.23(1.20) | −0.52[−1.08,0.03] | − |
| ダパグリフロジン錠5mg(n=41) | 8.49(1.16) | −0.47[−1.01,0.06] | 0.05(0.21) |
| ダパグリフロジン錠10mg(n=45) | 8.12(1.00) | −0.45[−0.96,0.05] | 0.07(0.21) |
| eGFR45以上60mL/min/1.73m2未満 | |||
| プラセボ(n=40) | 8.78(1.32) | −0.11[−0.57,0.35] | − |
| ダパグリフロジン錠5mg(n=35) | 8.13(0.93) | −0.47[−0.97,0.02] | −0.37(0.23) |
| ダパグリフロジン錠10mg(n=32) | 8.25(0.89) | −0.44[−0.94,0.07] | −0.33(0.24) |
17.2 製造販売後調査等
17.2.1 製造販売後臨床試験
(1)プラセボ対照二重盲検比較試験(DPP-4阻害薬との併用を含むインスリン製剤との併用療法、D1692C00013試験)
eGFRが45mL/min/1.73m2以上の2型糖尿病患者を対象とし、インスリン製剤〔0.2単位/kg/日以上かつ15単位/日以上〕の単独又はDPP-4阻害薬との併用療法に加え、ダパグリフロジン錠5mg併用16週間投与におけるHbA1c変化量の結果は以下のとおりであった。低血糖の有害事象発現割合は、16週間の二重盲検投与期ではダパグリフロジン錠併用群19.5%(24例/123例)、プラセボ併用群23.3%(14例/60例)であった。
| HbA1c(NGSP値)(%) | |||
| ベースライン平均値(SD) | ベースライン値からの変化量注4)(SE) | プラセボとの差(SE) | |
| プラセボ併用群(n=60) | 8.49(0.93) | 0.05(0.0904) | −0.60※(0.1053) |
| ダパグリフロジン錠併用群(n=122) | 8.26(0.79) | −0.55(0.0638) | |
18. 薬効薬理
18.1 作用機序
18.2 SGLT2に対する阻害作用
18.3 尿中グルコース排泄促進作用及び血糖低下作用
遺伝的糖尿病モデルのZDFラットにダパグリフロジンを単回経口投与した試験で、尿中グルコース排泄量の増加と共に血漿中グルコース濃度の低下が認められた42)。また、ZDFラットにダパグリフロジンを15日間反復経口投与した試験では、投与15日目の絶食下での尿中グルコース排泄量は用量依存的に増加し、投与8日目及び投与14日目にそれぞれ絶食下及び摂餌下での血漿中グルコース濃度は用量依存的に低下した43)。
日本人2型糖尿病患者を対象とした第I相反復投与試験において、ダパグリフロジン10mgを投与したとき、投与1及び14日目の投与後24時間までの累積尿中グルコース排泄量は増加し、投与13日目のOGTT後の血糖値のAUC0-4hrが低下した1)。
日本人2型糖尿病患者を対象とした第I相反復投与試験において、ダパグリフロジン10mgを投与したとき、投与1及び14日目の投与後24時間までの累積尿中グルコース排泄量は増加し、投与13日目のOGTT後の血糖値のAUC0-4hrが低下した1)。
19. 有効成分に関する理化学的知見
19.1. ダパグリフロジン
| 一般的名称 | ダパグリフロジン |
|---|---|
| 一般的名称(欧名) | Dapagliflozin |
| 化学名 | (1S)-1,5-Anhydro-1-C-{4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl}-D-glucitol |
| 分子式 | C21H25ClO6 |
| 分子量 | 408.87 |
| 物理化学的性状 | 白色〜微黄白色の粉末である。メタノールに溶けやすく、水に極めて溶けにくく、へプタンにほとんど溶けない。 |
| KEGG DRUG |
|
20. 取扱い上の注意
PTPシート又はプラスチックボトルから取り出した後は、高温・高湿を避けること。
22. 包装
<ダパグリフロジン錠5mg「EP」>
(PTP)100錠(10錠×10)
(プラスチックボトル:バラ:乾燥剤入り)300錠
<ダパグリフロジン錠10mg「EP」>
(PTP)100錠(10錠×10)
(プラスチックボトル:バラ:乾燥剤入り)300錠
23. 主要文献
- Kasichayanula S,et al., Diabetes Obes Metab., 13 (4), 357-365, (2011) »PubMed
- 国内第I相臨床試験−用量漸増単回投与試験(フォシーガ錠:2014年3月24日承認、CTD2.7.6.6.3)
- 生物学的同等性と食事の影響(フォシーガ錠:2014年3月24日承認、CTD2.7.6.4)
- 社内資料:生物学的同等性に関する資料
- Boulton DW,et al., Br J Clin Pharmacol., 75 (3), 763-768, (2013) »PubMed
- 蛋白結合率測定試験(フォシーガ錠:2014年3月24日承認、CTD2.6.5.6)
- Kasichayanula S,et al., Br J Clin Pharmacol., 76, 432-444, (2013) »PubMed
- Kasichayanula S,et al., Clin Ther., 33, 1798-1808, (2011) »PubMed
- 腎、肝、小腸ミクロソームによるグルクロン酸抱合(フォシーガ錠:2014年3月24日承認、CTD2.6.4.9)
- In vivo代謝(フォシーガ錠:2014年3月24日承認、CTD2.6.5.9)
- マスバランス及び代謝試験(フォシーガ錠:2014年3月24日承認、CTD2.7.6.9)
- 薬物動態学的薬物相互作用(フォシーガ錠:2014年3月24日承認、CTD2.6.4.7)
- 糞尿中排泄率(フォシーガ錠:2014年3月24日承認、CTD2.6.4.6)
- 腎機能障害患者における薬物動態(フォシーガ錠:2014年3月24日承認、CTD2.7.6.12)
- 肝機能障害患者における薬物動態(フォシーガ錠:2014年3月24日承認、CTD2.7.6.13)
- Kasichayanula S,et al., Diabetes Obes Metab., 13 (1), 47-54, (2011) »PubMed
- ピオグリタゾンとの薬物間相互作用試験(フォシーガ錠:2014年3月24日承認、CTD2.7.6.15)
- Imamura A,et al., Diabetes Ther., 4, 41-49, (2013) »PubMed
- ヒドロクロロチアジドとの薬物間相互作用試験(フォシーガ錠:2014年3月24日承認、CTD2.7.6.14)
- Wilcox CS,et al., J Am Heart Assoc., 7, e007046, (2018) »PubMed
- バルサルタン又はシンバスタチンとの薬物間相互作用試験(フォシーガ錠:2014年3月24日承認、CTD2.7.6.17)
- Kasichayanula S,et al., Adv Ther., 29 (2), 163-177, (2012) »PubMed
- Kasichayanula S,et al., Diabetes Obes Metab., 15 (3), 280-283, (2013) »PubMed
- 社内資料:溶出性に関する資料
- 国内試験(フォシーガ錠:2014年3月24日承認、CTD2.7.4.2.1.2.1)
- Kaku K,et al., Diabetes Obes Metab., 15 (5), 432-440, (2013) »PubMed
- 国内第IIb相試験(フォシーガ錠:2014年3月24日承認、CTD2.7.6.27)
- 単独療法プラセボ対照比較試験[1](フォシーガ錠:2014年3月24日承認、CTD2.7.6.28)
- Kaku K,et al., Diabetes Obes Metab., 16, 1102-1110, (2014) »PubMed
- 第III相単独投与比較試験(フォシーガ錠:2014年3月24日承認、CTD2.7.3.3)
- 単独療法プラセボ対照比較試験[2](フォシーガ錠:2014年3月24日 審査報告書)
- 単独又は併用療法による非盲検長期投与試験(フォシーガ錠:2014年3月24日承認、CTD2.7.6.48)
- Kaku K,et al., Diabetes Ther., 5, 415-433, (2014) »PubMed
- Kohan DE,et al., Kidney Int., 85 (4), 962-971, (2014) »PubMed
- Araki E,et al., J Diabetes Investig., 7, 555-564, (2016) »PubMed
- 2型糖尿病患者を対象としたインスリンとの併用療法試験(フォシーガ錠:2024年3月6日 再審査申請資料概要1.5.1.5)
- 2型糖尿病患者を対象としたインスリンとの併用療法試験(フォシーガ錠:2024年3月6日 再審査報告書)
- Araki E,et al., Diabetes Obes Metab., 19, 562-570, (2017) »PubMed
- Kanai Y,et al., J Clin Invest., 93, 397-404, (1994) »PubMed
- ヒトのSGLT2及びSGLT1に対するダパグリフロジンの阻害活性及び選択性(フォシーガ錠:2014年3月24日承認、CTD2.6.2.2.1.1)
- Wright EM,et al., J Intern Med., 261, 32-43, (2007) »PubMed
- 遺伝的糖尿病モデルZDFラットにおける単回投与試験(フォシーガ錠:2014年3月24日承認、CTD2.6.2.2.2.2)
- 糖尿病モデルZDFラットにおける反復投与試験(フォシーガ錠:2014年3月24日承認、CTD2.6.2.2.2.3)
24. 文献請求先及び問い合わせ先
文献請求先
第一三共エスファ株式会社
お客様相談室
〒103-0027
東京都中央区日本橋2-13-12
電話:0120-100-601
製品情報問い合わせ先
第一三共エスファ株式会社
お客様相談室
〒103-0027
東京都中央区日本橋2-13-12
電話:0120-100-601
26. 製造販売業者等
26.1 製造販売元
第一三共エスファ株式会社
東京都中央区日本橋本町3-5-1
26.2 販売提携
第一三共株式会社
東京都中央区日本橋本町3-5-1