医薬品情報
| 総称名 | レベトール |
|---|---|
| 一般名 | リバビリン |
| 欧文一般名 | Ribavirin |
| 薬効分類名 | 抗ウイルス剤 |
| 薬効分類番号 | 6250 |
| ATCコード | J05AP01 |
| KEGG DRUG |
D00423
リバビリン
|
| KEGG DGROUP |
DG03198
抗C型肝炎ウイルス薬
|
| JAPIC | 添付文書(PDF) |
添付文書情報2023年6月 改訂(第4版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| レベトールカプセル200mg | REBETOL Capsules 200mg | MSD | 6250022M1021 | 190.4円/カプセル | 劇薬, 処方箋医薬品 |
1. 警告
1.2 本剤では催奇形性及び遺伝毒性が報告されているので、妊娠する可能性のある女性に投与する場合には、本剤投与中及び最終投与後9ヵ月間において避妊するよう指導すること。[9.4.1、15.2.3参照]
1.3 本剤では催奇形性及び遺伝毒性が報告されており、本剤の精液中への移行が否定できないことから、パートナーが妊婦、妊娠している可能性又は妊娠する可能性のある男性に投与する場合には、本剤投与中及び最終投与後6ヵ月間においてバリア法(コンドーム)を用いて避妊するよう指導すること。[9.4.2、15.2.3参照]
2. 禁忌
次の患者には投与しないこと
2.2 本剤の成分又は他のヌクレオシドアナログ(アシクロビル、ガンシクロビル、ビダラビン等)に対し過敏症の既往歴のある患者
2.8 自己免疫性肝炎の患者[自己免疫性肝炎が悪化することがある。][11.1.4参照]
4. 効能または効果
○インターフェロン ベータとの併用による次のいずれかのC型慢性肝炎におけるウイルス血症の改善
(1)血中HCV RNA量が高値の患者
(2)インターフェロン製剤単独療法で無効の患者又はインターフェロン製剤単独療法後再燃した患者
5. 効能または効果に関連する注意
<併用薬剤共通>
5.1 C型慢性肝炎又はC型代償性肝硬変におけるウイルス血症の改善に対する本剤の併用にあたってはHCV RNAが陽性であること、及び組織像又は肝予備能、血小板数等により慢性肝炎又は代償性肝硬変であることを確認すること。
<インターフェロン ベータとの併用の場合>
5.2 血中HCV RNA量が高値のC型慢性肝炎に本剤を用いる場合、血中HCV RNA量がRT-PCR法で105IU/mL以上又はb-DNA法で1Meq./mL以上であることを確認すること。
<ソホスブビル・ベルパタスビル配合剤との併用の場合>
5.3 C型代償性肝硬変患者に対する治療は、ウイルス血症の改善を目的としたものであり、本併用療法によりウイルス学的効果が得られた場合であっても、肝硬変が治癒するものではないため、肝硬変に対する適切な処置は継続すること。
6. 用法及び用量
通常、成人には、下記の用法・用量のリバビリンを経口投与する。
本剤の投与に際しては、患者の状態を考慮し、減量、中止等の適切な処置を行うこと。
本剤の投与に際しては、患者の状態を考慮し、減量、中止等の適切な処置を行うこと。
<インターフェロン ベータ又はソホスブビル・ベルパタスビル配合剤との併用の場合>
| 患者の体重 | リバビリンの投与量 | ||
| 1日の投与量 | 朝食後 | 夕食後 | |
| 60kg以下 | 600mg | 200mg | 400mg |
| 60kgを超え80kg以下 | 800mg | 400mg | 400mg |
| 80kgを超える | 1,000mg | 400mg | 600mg |
7. 用法及び用量に関連する注意
<併用薬剤共通>
7.1 C型慢性肝炎又はC型代償性肝硬変におけるウイルス血症の改善に対する本剤の単独療法は無効である。
7.2 本剤の投与期間は、臨床効果(HCV RNA、ALT等)及び副作用の程度を考慮しながら慎重に決定すること。特に好中球数、血小板数、ヘモグロビン濃度の推移に注意し、本剤の減量あるいは中止基準に従うこと。
<インターフェロン ベータとの併用の場合>
7.3 インターフェロン ベータは、通常、成人には1日600万国際単位で投与を開始し、投与後4週間までは連日、以後週3回静脈内投与又は点滴静注する。
7.3.1 セログループ1[ジェノタイプI(1a)又はII(1b)]で血中HCV RNA量が高値の患者における通常の投与期間は48週間である。なお、24週間以上の投与で効果が認められない場合、投与の中止を考慮すること。[17.1.2参照]
7.3.2 それ以外の患者における通常の投与期間は24週間である。[17.1.1参照]
7.4 下表の臨床検査値を確認することが望ましい。国内臨床試験において、リバビリンとして体重あたり1日13mg/kgを超える量を投与した場合、貧血の発現頻度の増加が認められた。[8.2、10.2参照]
| 検査項目 | 投与前値 |
| 白血球数 | 4,000/mm3以上 |
| 血小板数 | 100,000/mm3以上 |
| ヘモグロビン濃度 | 12g/dL以上 |
7.5 投与中は、定期的に血液学的検査を実施し、好中球数、血小板数、ヘモグロビン濃度の低下が認められた場合には、下表を参考にして用量を変更すること。[2.3、8.2、9.1.2、10.2参照]
| 検査項目 | 数値 | 本剤 | インターフェロン ベータ |
| 白血球数 | 1,500/mm3未満 | 変更なし | 半量に減量 |
| 1,000/mm3未満 | 中止 | ||
| 好中球数 | 750/mm3未満 | 変更なし | 半量に減量 |
| 500/mm3未満 | 中止 | ||
| 血小板数 | 50,000/mm3未満 | 変更なし | 半量に減量 |
| 25,000/mm3未満 | 中止 | ||
| ヘモグロビン濃度 (心疾患又はその既往なし) | 10g/dL未満 | 減量 600mg/日→400mg/日 800mg/日→600mg/日 1,000mg/日→600mg/日 | 変更なし |
| 8.5g/dL未満 | 中止 | ||
| ヘモグロビン濃度 (心疾患又はその既往あり) | 10g/dL未満、又は投与中、投与前値に比べ2g/dL以上の減少が4週間持続 | 減量 600mg/日→400mg/日 800mg/日→600mg/日 1,000mg/日→600mg/日 | 変更なし |
| 8.5g/dL未満、又は減量後、4週間経過しても12g/dL未満 | 中止 | ||
<ソホスブビル・ベルパタスビル配合剤との併用の場合>
7.6 投与中は、定期的に血液学的検査を実施し、好中球数、血小板数、ヘモグロビン濃度の低下が認められた場合には、下表を参考にして用量を変更すること。
なお、投与を再開する場合には、臨床検査値が下表の中止基準を上回ったことを確認すること。また、血小板数の減少による投与中止後の本剤の再開は、下表を参考にすること。[2.3、9.1.2、10.2参照]
なお、投与を再開する場合には、臨床検査値が下表の中止基準を上回ったことを確認すること。また、血小板数の減少による投与中止後の本剤の再開は、下表を参考にすること。[2.3、9.1.2、10.2参照]
| 検査項目 | 数値 | 本剤 |
| 好中球数 | 500/mm3未満 | 中止 |
| 血小板数 | 50,000/mm3未満 | 中止 |
| 25,000/mm3未満 | 中止(再開不可) | |
| ヘモグロビン濃度 (心疾患又はその既往なし) | 10g/dL未満 | 減量 600mg/日→400mg/日 800mg/日→600mg/日 1,000mg/日→600mg/日 |
| 8.5g/dL未満 | 中止 | |
| ヘモグロビン濃度 (心疾患又はその既往あり) | 10g/dL未満、又は投与中、投与前値に比べ2g/dL以上の減少が4週間持続 | 減量 600mg/日→400mg/日 800mg/日→600mg/日 1,000mg/日→600mg/日 |
| 8.5g/dL未満、又は減量後、4週間経過しても12g/dL未満 | 中止 |
| 検査項目 | 数値 | 本剤 |
| 好中球数 | 500/mm3未満 | 中止 |
| 血小板数 | 50,000/mm3未満 | 中止 |
| 25,000/mm3未満 | 中止(再開不可) | |
| ヘモグロビン濃度 (心疾患又はその既往なし) | 投与開始1〜4週時 11g/dL未満 | 減量 600mg/日→200mg/日 800mg/日→400mg/日 1,000mg/日→400mg/日 |
| 投与開始5週時以降 10g/dL未満 | ||
| 8.5g/dL未満 | 中止 | |
| ヘモグロビン濃度 (心疾患又はその既往あり) | 投与開始1〜4週時 11g/dL未満、又は投与中、投与前値に比べ2g/dL以上の減少が4週間持続 | 減量 600mg/日→200mg/日 800mg/日→400mg/日 1,000mg/日→400mg/日 |
| 投与開始5週時以降 10g/dL未満、又は投与中、投与前値に比べ2g/dL以上の減少が4週間持続 | ||
| 8.5g/dL未満、又は減量後、4週間経過しても12g/dL未満 | 中止 |
8. 重要な基本的注意
<併用薬剤共通>
<インターフェロン ベータとの併用の場合>
8.2 ヘモグロビン濃度、白血球数、好中球数及び血小板数の検査は、投与開始後1週間は2〜3日に1回、以後投与開始後4週間までは毎週、その後は4週間に1回程度実施すること。[7.4、7.5、11.1.1-11.1.3、11.1.10、11.1.11参照]
8.4 抑うつ、自殺企図をはじめ、躁状態、攻撃的行動、不眠、不安、焦燥、興奮、攻撃性、易刺激性等の精神神経症状発現の可能性について患者及びその家族に十分理解させ、これらの症状があらわれた場合には直ちに連絡するよう注意を与えること。[2.6、9.1.6、11.1.6参照]
8.5 重篤な肝機能障害があらわれることがあるので、定期的に肝機能検査を行うなど観察を十分に行うこと。[11.1.3参照]
8.6 間質性肺炎があらわれることがあるので、咳嗽、呼吸困難等があらわれた場合には直ちに連絡するよう患者に対し注意を与えること。[11.1.8参照]
8.8 ネフローゼ症候群があらわれることがあるので、定期的に尿検査(尿蛋白)を行うなど観察を十分に行うこと。[11.1.11参照]
8.9 網膜症があらわれることがあるので、定期的に眼底検査を行うなど観察を十分に行うこと。また、視力低下、視野中の暗点が出現した場合は速やかに医師の診察を受けるよう患者を指導すること。[11.1.14参照]
8.11 溶血性尿毒症症候群があらわれることがあるので、定期的に血液検査(血小板数、赤血球数、末梢血液像等)及び腎機能検査を行うなど観察を十分に行うこと。[11.1.10参照]
<ソホスブビル・ベルパタスビル配合剤との併用の場合>
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
<インターフェロン ベータとの併用の場合>
9.1.1 投与開始前のヘモグロビン濃度が14g/dL未満あるいは好中球数が2,000/mm3未満の患者
<併用薬剤共通>
9.1.2 心疾患(ただしコントロールの困難な心疾患(心筋梗塞、心不全、不整脈等)を除く)又はその既往歴のある患者
9.1.3 痛風又はその既往歴のある患者
血清尿酸濃度の上昇が報告されている。
9.1.4 アレルギー素因のある患者
9.1.5 高度の白血球減少又は血小板減少のある患者
9.1.6 中枢・精神神経障害又はその既往歴のある患者
9.1.7 自己免疫疾患(ただし自己免疫性肝炎を除く)の患者又はその素因のある患者
9.1.8 高血圧症の患者
脳出血を含む脳血管障害が生じたとの報告がある。なお、高血圧症及び糖尿病の両疾患を合併する患者では脳出血が生じるリスクが高いので注意すること。[11.1.17参照]
9.1.9 糖尿病又はその既往歴、家族歴のある患者、耐糖能障害のある患者
9.2 腎機能障害患者
9.2.1 慢性腎不全又はクレアチニンクリアランスが50mL/分以下の腎機能障害のある患者
9.2.2 軽度又は中等度の腎機能障害のある患者(クレアチニンクリアランスが50mL/分以下の腎機能障害のある患者を除く)
本剤の血中濃度が上昇し、重大な副作用が生じることがある。[16.6.1参照]
9.3 肝機能障害患者
9.4 生殖能を有する者
9.4.1 妊娠する可能性のある女性
本剤投与中及び最終投与後9ヵ月間において避妊する必要性及び適切な避妊法について説明し、避妊するよう指導すること。また、投与直前の妊娠検査結果が陰性であることを確認後に投与を開始すること。なお、妊娠していないことを確認するために、妊娠検査を毎月1回実施すること。[1.1、1.2、9.5、15.2.3参照]
9.4.2 パートナーが妊婦、妊娠している可能性又は妊娠する可能性のある男性
本剤投与中及び最終投与後6ヵ月間においてバリア法(コンドーム)を用いて避妊するよう指導すること。精液中への本剤の移行が否定できないことから、その危険性を患者に十分理解させること。[1.3、9.5、15.2.3参照]
9.5 妊婦
9.6 授乳婦
授乳を避けさせること。動物実験(ラット)で乳汁中への移行が認められている。[2.1参照]
9.7 小児等
小児等に対する臨床試験は実施していない。
9.8 高齢者
患者の状態を観察しながら慎重に投与すること。国内で実施した臨床試験において、高齢者では、高度の臨床検査値異常等の発現頻度及び減量を要する頻度が高くなる傾向が認められている。
10. 相互作用
10.2 併用注意
| ヌクレオシドアナログ (ジダノシン、アバカビル硫酸塩等) | 併用により乳酸アシドーシス、肝不全が報告されていることから、本剤は乳酸アシドーシス、肝不全を増強する可能性がある。また、本剤投与終了後2ヵ月間はヌクレオシドアナログとの相互作用の可能性があるので注意すること。 | 本剤はin vitroにおいてプリンヌクレオシドのリン酸化を促進する。また、ジダノシンとの併用により、乳酸アシドーシス、膵炎など死亡例を含むミトコンドリア毒性の発現が報告されている。 |
| ジドブジン | 本剤はジドブジンの効果を減弱するおそれがある。併用する場合は、血漿中HIV RNAレベルを観察することが望ましい。HIV RNAレベルが上昇した場合には、本剤の中止等の適切な処置を行うこと。 | 本剤はin vitroにおいてジドブジンのリン酸化を阻害する。 |
| アザチオプリン [7.4-7.6参照] | 骨髄機能抑制が起こるおそれがある。併用する場合には、定期的に血液検査を行うなど、患者の状態を十分に観察すること。本剤の減量、中止については、「7.用法及び用量に関連する注意」の項を参照すること。 | 本剤がアザチオプリンの代謝酵素であるイノシン一リン酸脱水素酵素(IMPDH)を阻害することにより、代謝産物のメチルチオイノシン一リン酸(meTIMP)が蓄積すると考えられる。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
<インターフェロン ベータとの併用の場合>
11.1.1 貧血[赤血球減少(250万/mm3未満)(5%未満)、ヘモグロビン減少(8g/dL未満)(5%未満)、ヘモグロビン減少(8以上9.5g/dL未満)(5%以上)、ヘモグロビン減少(9.5以上11g/dL未満)(5%以上)][2.3、2.4、8.1、8.2、9.1.2参照]
11.1.3 重篤な肝障害(5%未満)
11.1.5 脳梗塞(5%未満)
11.1.6 重篤なうつ状態、自殺企図、躁状態、攻撃的行動(いずれも頻度不明)
11.1.7 せん妄、幻覚(いずれも頻度不明)
11.1.8 間質性肺炎(頻度不明)
発熱、咳嗽、呼吸困難等の呼吸器症状、また、胸部X線異常があらわれた場合には投与を中止し、副腎皮質ホルモン剤の投与等の適切な処置を行うこと。[8.6参照]
11.1.9 心不全(頻度不明)[2.3参照]
11.1.10 溶血性尿毒症症候群(HUS)(頻度不明)
11.1.11 ネフローゼ症候群(頻度不明)
11.1.12 糖尿病(1型及び2型)(頻度不明)
11.1.13 敗血症(頻度不明)
易感染性となり、敗血症があらわれることがある。[9.1.5参照]
11.1.14 網膜症(頻度不明)
網膜出血、軟性白斑及び糖尿病網膜症の増悪に注意すること。[8.9参照]
<ソホスブビル・ベルパタスビル配合剤との併用の場合>
11.2 その他の副作用
| 5%以上 | 5%未満 | 頻度不明 | |
| 全身症状 | 発熱注1)、悪寒(82.2%)、全身倦怠感(88.5%)、かぜ症候群 | インフルエンザ様症状 | |
| 過敏症 | 発疹、そう痒感 | 蕁麻疹 | |
| 血液 | 白血球数減少(75.3%)、血小板数減少(62.1%)、顆粒球数減少(81.6%)、白血球分画異常(96.6%)、赤血球数減少(70.7%)、ヘモグロビン減少(76.4%)、ヘマトクリット減少(71.3%)、網状赤血球数減少、網状赤血球数増多(75.9%)、好酸球数増多、好中球数増多、血小板数増多 | 出血傾向、白血球数増多 | |
| 肝臓 | AST上昇、ALT上昇、Al-P上昇、LDH上昇、総ビリルビン上昇、γ-GTP上昇 | ||
| 腎臓 | 蛋白尿(50.6%)、BUN上昇、血尿 | クレアチニン上昇、膀胱炎、頻尿、排尿障害 | |
| 精神神経系 | 頭痛・頭重(80.5%)、不眠、めまい、抑うつ、焦燥、手足のしびれ、不安 | 意識障害、傾眠、知覚異常、振戦、無気力、歩行困難、健忘、異常感、感情不安定、耳閉、注意力障害 | 妄想、怒り |
| 循環器 | 血圧上昇、動悸、潮紅、四肢冷感 | 不整脈、血圧低下 | |
| 呼吸器 | 咳嗽、上気道炎、呼吸困難、鼻出血 | 肺炎、鼻漏、血痰、嗄声、鼻炎、気管支炎、鼻閉 | |
| 消化器 | 食欲不振(59.2%)、悪心・嘔吐、下痢、腹痛、消化不良、便秘、口内・口唇炎、味覚異常 | 腹部膨満感、口渇、歯周・歯髄・歯肉炎、歯痛、胃炎、歯の異常、排便障害、腸炎、舌炎、痔核、おくび、鼓腸放屁、腸管機能異常 | 膵炎 |
| 皮膚 | 湿疹、脱毛 | ざ瘡、発汗、皮膚乾燥、白癬、紅斑、紫斑、脂漏、爪の異常、過角化、皮膚潰瘍、毛質異常、落屑 | 丘疹 |
| 眼 | 眼底出血等の網膜の微小循環障害注2) | 眼痛、視力異常、結膜下出血、眼球充血、結膜炎、眼の異和感、眼そう痒症、眼精疲労、硝子体浮遊物、羞明、視覚異常、視野欠損、麦粒腫 | |
| 注射部位 | 発赤 | 疼痛、熱感、腫脹、色素沈着、そう痒、出血 | |
| その他 | 関節痛(58.0%)、筋肉痛、肩こり等の緊張亢進、背部・腰部痛、浮腫、胸部圧迫感、疼痛、咽頭炎、体重減少、尿糖、血清アルブミン低下(54.0%)、血清総蛋白減少、血清コレステロール上昇、血中コレステロール低下、血中尿酸上昇、血清カルシウム低下、血清無機リン低下、CRP上昇 | 疲労、脱力感、難聴、単純疱疹、帯状疱疹、蜂窩織炎、筋痙直、手指関節拘縮、耳鳴、冷汗、不正出血、神経痛、頚部痛、易感染性、花粉症、外耳炎、耳痛、中耳炎、前立腺炎、嗅覚異常、四肢不快感、サルコイドーシス、トリグリセライド上昇、血清アミラーゼ上昇、血糖上昇 | CK上昇、血清カリウム上昇、ヘモグロビンA1C上昇 |
| 5〜10%未満 | 5%未満 | 頻度不明 | |
| 感染 | 咽頭炎 | ||
| 神経系 | 頭痛 | ||
| 循環器 | 徐脈 | ||
| 消化器 | 悪心、口内炎 | ||
| 皮膚及び皮下組織 | そう痒症、発疹 | 血管性浮腫 | |
| その他 | 倦怠感 |
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
15. その他の注意
15.2 非臨床試験に基づく情報
15.2.1 マウス3及び6ヵ月間投与試験(1〜150mg/kg/日)で精子異常(15mg/kg/日以上)がみられたとの報告がある(休薬により回復)。
15.2.2 ラット長期投与試験(24ヵ月間、10〜40mg/kg/日)で網膜変性の発生頻度が対照群に比べて増加したとの報告がある。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
健康成人男性(6名)に本剤200、400、600、800、1,000及び1,200mgを空腹時に単回経口投与したとき、血漿中未変化体濃度のCmaxについては200〜800mg、AUC0-tについては200〜1,000mgの用量範囲でそれぞれ線形性が認められ、それ以上の投与量では吸収の頭打ちが示唆された1)。
16.1.2 反復投与
C型慢性肝炎患者(15名)に本剤400mg(800mg/日)を朝夕食後に1日2回48週間、ペグインターフェロン アルファ-2b(遺伝子組換え)(以下:PEG-IFNα-2b)の1.5μg/kg週1回皮下投与との併用により、反復経口投与したときの血清中未変化体濃度を以下の図表に示した。血清中未変化体濃度は投与開始後8週目までに定常状態に到達し、Cmax、Cmin及びAUC0-12hrに基づく累積係数はそれぞれ6.53、12.2及び9.42であった。定常状態に到達後の消失半減期は286時間であった2)。
図1 C型慢性肝炎患者における血清中濃度(平均値±標準偏差)
| tmax(hr) | Cmax(μg/mL) | Cmin(μg/mL) | AUC0-12hr†(μg・hr/mL) | t1/2(hr) | CL/F(L/hr) | Vd/F(L) | |
| 定常状態(N=14)†† | 3.00 | 3.33 | 2.42 | 32.5 | 286 | 12.7§ | 5374§ |
| 初回投与(N=15) | 3.33 | 0.604 | 0.221 | 4.02 | 27.1 | 37.8 | 1472 |
| 累積係数 | 6.53§ | 12.2§ | 9.42§ |
16.1.3 静脈内投与時
健康成人男性(6名)にリバビリン溶液150mgを急速静脈内投与したとき、血漿中未変化体の全身クリアランス(CL)は40.5L/hr、定常状態における見かけの分布容積(Vss)は241Lであった。同一被験者に本剤400mgを空腹時に経口投与したときのAUCとの比較によって算出した絶対バイオアベイラビリティ(経口投与時のAUC/静脈内投与時のAUC)は64%であった4)(外国人データ)。
16.2 吸収
16.2.1 食事の影響
健康成人男女(17名)に本剤600mgを食後又は空腹時に単回経口投与したとき、食後投与時ではCmax及びAUCが約70%上昇し、tmaxの遅延が認められた3)(外国人データ)。
16.3 分布
16.3.1 血漿蛋白結合
ヒト血漿蛋白と本薬との結合は全く認められず、非結合率はほぼ100%であった7)(in vitro)。
16.3.2 血球移行
健康成人男性(6名)に14C-標識リバビリンカプセル604mgを空腹時に単回経口投与したとき、赤血球中放射能濃度は血液(全血)中放射能濃度の約2倍の値を示したことから、血中放射性成分の大部分は赤血球中に存在しているものと推察された8)(外国人データ)。
16.3.3 組織内分布
ラットに14C-標識リバビリン溶液20mg/kgを1日1回21日間反復経口投与したとき、組織中放射能濃度は血球を除く殆どの組織で投与7日目までに定常状態に到達し、全身組織への広範な放射能分布が認められた。組織中放射能濃度は肝臓で最も高く、次いで腎臓、心臓、筋肉、肺、脾臓、膵臓、腸間膜リンパ節、前立腺、膀胱、骨髄に高濃度に分布した9)。
16.3.4 胎盤・胎児移行
妊娠ラットに14C-標識リバビリン溶液20mg/kgを単回経口投与したとき、胎児組織中への放射能の移行が認められた10)。
16.4 代謝
本剤の体内からの消失に関わる主要な代謝経路は、ribofuranosyl基の脱離及び3位側鎖(carboxamide)の加水分解であり、代謝物として1H-1,2,4-triazole-3-carboxamide(TCONH2)、1-β-D-ribofuranosyl-1H-1,2,4-triazole-3-carboxylic acid(RTCOOH)及び1H-1,2,4-triazole-3-carboxylic acid(TCOOH)が確認されている。本剤の薬効に関与しているもう一つの代謝経路は、ribofuranosyl基5'位のリン酸化であり、代謝物としてリバビリン一リン酸(RMP)、リバビリン二リン酸(RDP)及びリバビリン三リン酸(RTP)が確認されている。これらのリン酸化体は組織細胞中にのみ存在し、細胞外(血漿、尿)には認められない。
ヒト肝ミクロソームを用いたin vitro代謝実験の結果、上記のいずれの代謝経路についても、チトクロムP450系の介在は否定されている8)11)12)13)。
ヒト肝ミクロソームを用いたin vitro代謝実験の結果、上記のいずれの代謝経路についても、チトクロムP450系の介在は否定されている8)11)12)13)。
16.5 排泄
16.5.1 尿・糞中排泄
健康成人男性(6名)に14C-標識リバビリンカプセル604mgを空腹時に単回経口投与したとき、投与後14日目までの尿及び糞中放射能排泄率はそれぞれ61%及び12%であった。同時点までの尿中未変化体排泄率は投与量の17%であり、尿中放射能に占める割合は約27%であった8)(外国人データ)。
16.5.2 胆汁中排泄
ラットに14C-標識リバビリン溶液20mg/kgを単回経口投与したとき、投与後48時間までの胆汁中放射能排泄率は投与量の0.8%未満であった14)。
16.5.3 乳汁中への移行
授乳中のラットに14C-標識リバビリン溶液20mg/kgを単回経口投与したとき、放射能濃度の母乳/血漿比は0.6〜1.3であり、本薬又は代謝物の乳汁中への移行性が認められた15)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
腎機能障害患者(18名)に本剤400mgを空腹時に単回経口投与したときの血漿中未変化体濃度のパラメータを下表に示した。腎機能障害患者では、クレアチニンクリアランスに応じた全身クリアランス(CL/F)の低下が認められている6)(外国人データ)。[2.5、9.2.1、9.2.2参照]
| CLcr(mL/分) | 患者数 | Cmax(μg/mL) | AUC0-t(μg・hr/mL) | CL/F(L/hr) | CLr(L/hr) |
| ≧90 | 6名 | 0.630 | 9.65 | 53.2 | 7.74 |
| 61〜90 | 6名 | 0.821 | 17.5 | 29.8 | 4.31 |
| 31〜60 | 6名 | 0.732 | 20.4 | 24.2 | 2.15 |
| 10〜30 | 6名 | 1.16 | 31.7 | 13.0 | 0.696 |
人工透析依存の腎不全患者(6名)に本剤400mgを空腹時に単回経口投与したとき、人工透析クリアランス(CLhd=4.04L/hr)はクレアチニンクリアランスが61〜90(mL/分)の腎機能障害患者の腎クリアランス(4.31L/hr)にほぼ相当する値であったが、血漿中未変化体濃度について人工透析による明らかな変化は認められなかった(外国人データ)。[2.5参照]
16.6.2 肝機能障害患者
16.7 薬物相互作用
16.7.1 チトクロムP450系への影響
ヒト肝ミクロソームを用いたin vitro阻害実験の結果、CYP3A4、2D6、1A2、2E1、2C9/10の各P450分子種についてリバビリン添加による阻害作用は認められなかった13)。
ラットにリバビリン溶液を1日1回7日間反復経口投与したとき、120mg/kgまでの投与量では肝薬物代謝酵素系への誘導作用は認められなかった16)。
16.7.2 PEG-IFNα-2bの影響
C型慢性肝炎患者(12〜17名)を対象とした本剤600〜1,200mg/日の1日2回経口投与とPEG-IFNα-2b 0.35、0.7又は1.4μg/kg週1回皮下投与との併用による4週間反復投与試験において、薬物動態学的相互作用を示唆する所見は認められなかった17)(外国人データ)。
16.7.3 水酸化マグネシウム・水酸化アルミニウム併用の影響
健康成人男女(12名)に本剤600mgを空腹時に単独又は水酸化マグネシウム・水酸化アルミニウム含有製剤と併用したとき、併用時ではCmax、AUC0-tがそれぞれ3.3%、13.7%減少したが、Tmaxに影響は認められなかった18)(外国人データ)。
(注)
本剤の承認された1日投与量は600〜1,000mgである。
PEG-IFNα-2b製剤は承認整理済である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
<インターフェロン ベータとの併用の場合>
17.1.1 国内第III相試験(24週間投与での成績)
「セログループ1(ジェノタイプ1)かつ血中HCV RNA量が高値の患者:対象A」及び「セログループ1(ジェノタイプ1)以外かつ血中HCV RNA量が高値の患者、及びインターフェロン(以下:IFN)製剤による治療歴のある血中HCV RNA量が低値の患者:対象B」を対象として、インターフェロン ベータ(以下:IFNβ)1日6.0×106IUを4週間連日投与後、1日6.0×106IUを週3回20週間投与し、本剤1日600〜800mgを24週間併用投与した際の、IFNβ/本剤併用群と対照薬群であるインターフェロン アルファ-2b(遺伝子組換え)(以下:IFNα-2b)/本剤併用群におけるセログループ別(ジェノタイプ別)及びウイルス量(アンプリコア法)別の投与終了後24週目のHCV RNA陰性化率(アンプリコア法)は、下記のとおりであった。
| HCV RNA陰性化率 | ||
| IFNβ+本剤併用群 | IFNα-2b+本剤併用群 | |
| 対象A | 18.7%(17/91) | 15.6%(7/45) |
| 対象B | 80.0%(20/25) | 83.3%(10/12) |
116例全例に副作用(臨床検査値異常を含む)が認められた。主な副作用は、発熱(98.3%)、倦怠感(89.7%)、頭痛(81.0%)、悪寒(81.0%)、食欲不振(61.2%)等であり、臨床検査値の異常は、好中球数減少(78.4%)、網状赤血球数増加(78.4%)、ヘモグロビン減少(75.0%)、白血球数減少(74.1%)、リンパ球百分率増加(71.6%)等であった。
17.1.2 国内第III相試験(48週間投与での成績)
セログループ1(ジェノタイプ1)で血中HCV RNA量が高値であり、うつ病の既往歴(インターフェロン アルファ製剤によるうつ病の既往歴を含む)のあるC型慢性肝炎患者、又はうつ病の合併症あるいはその疑いのあるC型慢性肝炎患者(ハミルトンうつ病評価尺度17項目の総スコアが13以下)を対象として、IFNβ1日6.0×106IUを4週間連日投与後、1日6.0×106IUを週3回44週間投与し、本剤1日400〜1,000mgを48週間併用投与する試験を精神科医による診察を定期的に行った上で実施した。投与開始24週後、48週後の投与中止率はそれぞれ8.6%(5/58)、17.2%(10/58)であり、48週間投与終了後24週目のHCV RNA陰性化率(アンプリコア法)は22.4%(13/58)であった。
58例全例に副作用(臨床検査値異常を含む)が認められた。主な副作用は、発熱(98.3%)、倦怠感(86.2%)、悪寒(84.5%)、頭痛(79.3%)、食欲不振(55.2%)等であり、臨床検査値の異常は、好中球数減少(81.0%)、ヘマトクリット減少(79.3%)、ヘモグロビン減少(79.3%)、白血球数減少(77.6%)、赤血球数減少(74.1%)等であった。
<ソホスブビル・ベルパタスビル配合剤との併用の場合>
17.1.3 国内第III相臨床試験
C型肝炎直接型抗ウイルス薬(DAA)による治療歴を有するジェノタイプ1又は2のC型慢性肝炎又はC型代償性肝硬変患者を対象として、本剤とソホスブビル・ベルパタスビル配合剤併用時の有効性及び安全性を検討することを目的とした第III相臨床試験(無作為化非盲検並行群間比較試験)を実施した(12週間又は24週間投与)。主要評価項目はSVR12率であった。本剤及びソホスブビル・ベルパタスビル配合剤の24週間併用投与群の結果を表5に示す。
| 対象 | SVR12率 | |
| 全体 | 96.7%(58/60例) | |
| 年齢 | 65歳未満 | 96.8%(30/31例) |
| 65歳以上 | 96.6%(28/29例) | |
| HCVジェノタイプ | ジェノタイプ1 | 97.9%(47/48例) |
| ジェノタイプ2 | 91.7%(11/12例) | |
| 代償性肝硬変注) | なし | 94.9%(37/39例) |
| あり | 100.0%(21/21例) | |
| DAA治療歴 | NS5A阻害剤+NS3/4Aプロテアーゼ阻害剤 | 97.4%(37/38例) |
| NS5A阻害剤+NS5Bポリメラーゼ阻害剤 | 100.0%(8/8例) | |
| NS5Bポリメラーゼ阻害剤単独 | 100.0%(8/8例) | |
| NS5A阻害剤+NS3/4Aプロテアーゼ阻害剤+NS5Bポリメラーゼ阻害剤 | 100.0%(5/5例) | |
| NS5Bポリメラーゼ阻害剤+NS3/4Aプロテアーゼ阻害剤 | 0%(0/1例) | |
60例中21例(35.0%)に副作用が認められた。主な副作用は、貧血13例(21.7%)、倦怠感3例(5.0%)、そう痒症2例(3.3%)であった。
(注)IFNα-2b製剤及びPEG-IFNα-2b製剤は承認整理済である。
18. 薬効薬理
18.1 作用機序
18.1.1 抗HCV作用
18.1.2 抗ウイルス作用機序
リバビリンの詳細な作用機序は明らかでない。リバビリンは細胞内でリン酸化され、HCV由来RNA依存性RNAポリメラーゼによるグアノシン三リン酸のRNAへの取込みを抑制した(in vitro)。また、HCV由来RNA依存性RNAポリメラーゼによるRNA生成過程でリバビリン三リン酸がRNAに取り込まれ、このことがウイルスのゲノムを不安定にすると考えられた(in vitro)20)。
19. 有効成分に関する理化学的知見
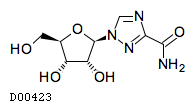
19.1. リバビリン
| 一般的名称 | リバビリン |
|---|---|
| 一般的名称(欧名) | Ribavirin |
| 化学名 | 1-β-D-Ribofuranosyl-1H-1,2,4-triazole-3-carboxamide |
| 分子式 | C8H12N4O5 |
| 分子量 | 244.20 |
| 融点 | 167-171℃ |
| 物理化学的性状 | 白色の結晶性の粉末である。水又はN,N-ジメチルホルムアミドに溶けやすく、メタノールに溶けにくく、エタノール(99.5)にほとんど溶けない。結晶多形が認められる。 |
| 分配係数 | (1-オクタノール−水系) pH2:3.76×10−3 pH4:3.85×10−3 pH6:3.44×10−3 pH8:1.38×10−3 pH10:1.70×10−4 pH12:1.78×10−4 |
| KEGG DRUG |
|
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
28カプセル[14カプセル(PTP)×2]
23. 主要文献
- 深瀬広幸ほか, 臨床医薬, 18, 521-38, (2002)
- 社内資料:反復投与(2004年10月22日承認、CTD2.7.2.2)
- 社内資料:食事の影響(2001年11月21日承認、申請資料概要ヘ.4.(5))
- 社内資料:バイオアベイラビリティ(2001年11月21日承認、申請資料概要ヘ.4.(4))
- 社内資料:肝機能障害患者(2001年11月21日承認、申請資料概要ヘ.4.(8))
- 社内資料:腎機能障害患者(2001年11月21日承認、申請資料概要ヘ.4.(9))
- 社内資料:血漿蛋白結合率(2001年11月21日承認、申請資料概要ヘ.3.(2).3))
- 社内資料:ヒトにおける体内動態(2001年11月21日承認、申請資料概要ヘ.4.(1))
- 社内資料:組織内分布(2001年11月21日承認、申請資料概要ヘ.3.(2).1))
- 社内資料:胎盤・胎児移行性(2001年11月21日承認、申請資料概要ヘ.3.(2).2))
- 社内資料:代謝:ラット(2001年11月21日承認、申請資料概要ヘ.3.(3).1).[1])
- 社内資料:代謝:サル(2001年11月21日承認、申請資料概要ヘ.3.(3).1).[2])
- 社内資料:チトクロムP450に対する阻害作用(2001年11月21日承認、申請資料概要ヘ.3.(3).2).[1])
- 社内資料:胆汁中排泄(2001年11月21日承認、申請資料概要ヘ.3.(4).2))
- 社内資料:乳汁移行性(2001年11月21日承認、申請資料概要ヘ.3.(4).3))
- 社内資料:肝薬物代謝酵素誘導(2001年11月21日承認、申請資料概要ヘ.3.(3).2).[2])
- Glue P,et al., Hepatology., 32, 647-53, (2000) »PubMed
- 社内資料:制酸剤の影響(2001年11月21日承認、申請資料概要ヘ.4.(6))
- 社内資料:レプリコンを用いたリバビリンの評価試験
- 社内資料:リバビリンの抗HCV作用機序(2001年11月21日承認、申請資料概要ホ.1.(2).7))
- 社内資料:抗ウイルス作用を裏付ける試験(2004年10月22日承認、CTD2.6.2.2)
24. 文献請求先及び問い合わせ先
文献請求先
MSD株式会社
MSDカスタマーサポートセンター
東京都千代田区九段北1-13-12
電話:医療関係者の方:フリーダイヤル0120-024-961
製品情報問い合わせ先
MSD株式会社
MSDカスタマーサポートセンター
東京都千代田区九段北1-13-12
電話:医療関係者の方:フリーダイヤル0120-024-961
26. 製造販売業者等
26.1 製造販売元
MSD株式会社
東京都千代田区九段北1-13-12