医薬品情報
| 総称名 | ギャバロン |
|---|---|
| 一般名 | バクロフェン |
| 欧文一般名 | Baclofen |
| 製剤名 | バクロフェン髄注 |
| 薬効分類名 | 抗痙縮剤 |
| 薬効分類番号 | 1249 |
| ATCコード | M03BX01 |
| KEGG DRUG |
D00241
バクロフェン
|
| JAPIC | 添付文書(PDF) |
添付文書情報2025年2月 改訂(第2版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ギャバロン髄注0.005% | GABALON INTRATHECAL INJECTION | 第一三共 | 1249401A1022 | 1159円/管 | 劇薬, 処方箋医薬品注) |
| ギャバロン髄注0.05% | GABALON INTRATHECAL INJECTION | 第一三共 | 1249401A2029 | 23045円/管 | 劇薬, 処方箋医薬品注) |
| ギャバロン髄注0.2% | GABALON INTRATHECAL INJECTION | 第一三共 | 1249401A3025 | 23012円/管 | 劇薬, 処方箋医薬品注) |
1. 警告
1.1 本剤の長期持続投与は、本剤の髄腔内持続投与用に承認された専用のポンプシステムと組み合わせて行うため、ポンプシステムの植込み手術ならびに専用機器による用量の調節を伴う。したがって、本剤の長期持続投与は、当該手技及び専用機器の取り扱いに関する講習を受けた上で、本剤の安全性及び有効性を十分理解し、施術に関する十分な知識・経験のある医師のみが行うこと。
1.2 本剤の長期連用中に投与が突然中断されると離脱症状(高熱、精神状態の変化、強いリバウンド痙縮、筋硬直、横紋筋融解症等)が発現し、死亡に至る例も報告されているので、「使用上の注意」に十分留意し、離脱症状が発現しないよう適切な措置を講じるとともに、患者に対し離脱症状発現の可能性について十分説明すること。[7.3、8.1参照]
1.3 本剤の投与に際しては、患者又はそれに代わり得る適切な者に対して、本剤の危険性、本剤の投与が長期にわたる可能性があること、ならびに長期持続投与時には専用のポンプシステムと組み合わせて使用する必要があり、ポンプシステムに由来する危険性があることを十分に説明し、文書による同意を得た上で投与を開始すること。
2. 禁忌
次の患者には投与しないこと
2.1 本剤の成分に対し過敏症の既往歴のある患者
2.2 ポンプシステム植込み前に感染症に罹患している患者[感染症に罹患している患者では、術後の合併症のリスクが高まるため。]
4. 効能または効果
脳脊髄疾患に由来する重度の痙性麻痺(既存治療で効果不十分な場合に限る)
6. 用法及び用量
<髄注0.005%>
スクリーニング[効果の確認]
本剤専用のポンプシステムを植込む前に本剤の効果を確認するため、スクリーニングを実施する。スクリーニングには髄注0.005%(0.05mg/1mL)を用いる。
通常、成人にはバクロフェンとして1日1回50μg[髄注0.005%を1mL(1管)]をバルボタージ法(ポンピング)により髄腔内投与し、抗痙縮効果を1〜8時間後に確認する。期待した効果が認められない場合、初回投与から24時間以降に75μg[髄注0.005%を1.5mL(1.5管)]に増量の上同様に髄腔内投与して1〜8時間後に効果を確認する。期待した効果が認められない場合、2回目の投与から24時間以降に100μg[髄注0.005%を2mL(2管)]に増量の上同様に髄腔内投与して1〜8時間後に効果を確認する。100μgでも効果が認められない場合、本剤の治療対象とはならない。
通常、小児にはバクロフェンとして1日1回25μg[髄注0.005%を0.5mL(0.5管)]をバルボタージ法(ポンピング)により髄腔内投与し、抗痙縮効果を1〜8時間後に確認する。ただし、体格、症状などを考慮して増量することができるが、初回投与量の上限は50μg[髄注0.005%を1mL(1管)]とする。期待した効果が認められない場合、初回投与量が50μg未満である場合は50μg、50μgである場合は75μgに増量の上、髄腔内投与して1〜8時間後に効果を確認する。期待した効果が認められない場合、成人の用法・用量に準じて増量の上、同様に髄腔内投与して1〜8時間後に効果を確認する。100μgでも効果が認められない場合、本剤の治療対象とはならない。
<髄注0.05%>
適正用量の設定
本剤専用のポンプシステム植込み後の適正用量の設定には、髄注0.05%(10mg/20mL)または髄注0.2%(10mg/5mL)を用いる。髄注0.2%は0.05〜0.2%の範囲内で日局生理食塩液にて希釈して使用することができる。
1.用量設定期(滴定期)[ポンプシステム植込み後60日まで]
スクリーニングのいずれかの用量で期待した抗痙縮効果が認められた患者には、その用量を初回1日用量とし、本剤専用の植込み型ポンプシステムを用い24時間かけて髄腔内投与する。
通常、成人には1日用量が50〜250μgとなる範囲で患者の症状に応じ適宜増減する。用量の調整は通常1日に1回、次のとおりとする。なお、1日用量の上限は600μgとする。
| 原疾患 | 増量時 | 減量時 |
| 脊髄疾患(脊髄損傷、脊髄小脳変性症(痙性対麻痺)等) | 30%以内の範囲 | 20%以内の範囲 |
| 脳疾患(脳性麻痺、頭部外傷等) | 15%以内の範囲 | 20%以内の範囲 |
通常、小児には1日用量が25〜150μgとなる範囲で患者の症状に応じ適宜増減する。用量の調整は通常1日に1回、次のとおりとする。なお、1日用量の上限は400μgとする。
| 増量時 | 減量時 | |
| 小児 | 15%以内の範囲 | 20%以内の範囲 |
2.維持期[ポンプシステム植込み後61日以降]
通常、成人では標準1日用量として50〜250μgであるが、患者の本剤に対する反応には個人差があるため、症状に応じて適宜増減する。用量の調整は通常1日に1回、次のとおりとする。なお、1日用量の上限は600μgとする。
| 原疾患 | 増量時 | 減量時 |
| 脊髄疾患(脊髄損傷、脊髄小脳変性症(痙性対麻痺)等) | 40%以内の範囲 | 20%以内の範囲 |
| 脳疾患(脳性麻痺、頭部外傷等) | 20%以内の範囲 | 20%以内の範囲 |
通常、小児では標準1日用量として25〜150μgであるが、患者の本剤に対する反応には個人差があるため、症状に応じて適宜増減する。用量の調整は通常1日に1回、次のとおりとする。なお、1日用量の上限は400μgとする。
| 増量時 | 減量時 | |
| 小児 | 20%以内の範囲 | 20%以内の範囲 |
<髄注0.2%>
適正用量の設定
本剤専用のポンプシステム植込み後の適正用量の設定には、髄注0.05%(10mg/20mL)または髄注0.2%(10mg/5mL)を用いる。髄注0.2%は0.05〜0.2%の範囲内で日局生理食塩液にて希釈して使用することができる。
1.用量設定期(滴定期)[ポンプシステム植込み後60日まで]
スクリーニングのいずれかの用量で期待した抗痙縮効果が認められた患者には、その用量を初回1日用量とし、本剤専用の植込み型ポンプシステムを用い24時間かけて髄腔内投与する。
通常、成人には1日用量が50〜250μgとなる範囲で患者の症状に応じ適宜増減する。用量の調整は通常1日に1回、次のとおりとする。なお、1日用量の上限は600μgとする。
| 原疾患 | 増量時 | 減量時 |
| 脊髄疾患(脊髄損傷、脊髄小脳変性症(痙性対麻痺)等) | 30%以内の範囲 | 20%以内の範囲 |
| 脳疾患(脳性麻痺、頭部外傷等) | 15%以内の範囲 | 20%以内の範囲 |
通常、小児には1日用量が25〜150μgとなる範囲で患者の症状に応じ適宜増減する。用量の調整は通常1日に1回、次のとおりとする。なお、1日用量の上限は400μgとする。
| 増量時 | 減量時 | |
| 小児 | 15%以内の範囲 | 20%以内の範囲 |
2.維持期[ポンプシステム植込み後61日以降]
通常、成人では標準1日用量として50〜250μgであるが、患者の本剤に対する反応には個人差があるため、症状に応じて適宜増減する。用量の調整は通常1日に1回、次のとおりとする。なお、1日用量の上限は600μgとする。
| 原疾患 | 増量時 | 減量時 |
| 脊髄疾患(脊髄損傷、脊髄小脳変性症(痙性対麻痺)等) | 40%以内の範囲 | 20%以内の範囲 |
| 脳疾患(脳性麻痺、頭部外傷等) | 20%以内の範囲 | 20%以内の範囲 |
通常、小児では標準1日用量として25〜150μgであるが、患者の本剤に対する反応には個人差があるため、症状に応じて適宜増減する。用量の調整は通常1日に1回、次のとおりとする。なお、1日用量の上限は400μgとする。
| 増量時 | 減量時 | |
| 小児 | 20%以内の範囲 | 20%以内の範囲 |
7. 用法及び用量に関連する注意
7.1 バクロフェンの髄腔内及び経口以外の投与経路におけるヒトでの薬物動態、有効性及び安全性は国内においては確認されていないため、静脈内、筋肉内、皮下又は硬膜外への投与は行わないこと。
7.2 髄注0.005%は、スクリーニング専用の製剤であり、適正用量の設定には用いないこと。髄注0.05%及び髄注0.2%は、専用のポンプシステムと組み合わせて適正用量の設定に使用する製剤であり、スクリーニングには使用しないこと。
7.3 用量を調整する際には、用法及び用量に従うこと。適切な手順に従わなかったり、使用する薬液濃度を誤った場合、離脱症状や過量投与が発現するおそれがあるため、注意すること。[1.2、8.1、13.参照]
7.4 本剤の中止に際しては、1日用量の20%以内の範囲で2日ごとに減量し、患者の状態を慎重に観察しながらポンプシステム植込み時の初回1日用量まで減量すること。なお、本剤の投与再開に際しては、用量設定期における初回投与量から開始し、用量の増減については用量設定期の用法及び用量に従うこと。
7.5 投薬中の経口抗痙縮薬は、患者の状態を慎重に観察しながら、本剤による治療開始前又は治療開始後の適切な時期に減量又は漸次中止を試みること。ただし、急激な減量又は中止を避けること。
7.6 臨床試験では、カテーテル先端を第10胸椎(T10)以下に設置して本剤が投与されており、より高位に留置した場合には、呼吸抑制等の重篤な副作用が発現するおそれがあるので注意すること。
7.7 体躯が極端に小さい患者の場合には、通常よりも低用量からスクリーニング試験を開始することを考慮すること。
7.8 スクリーニング実施時及びポンプシステム植込み直後の用量設定期には、過量投与など重篤な副作用発現に備え、注意深く観察するとともに蘇生設備を確保しておくこと。
7.9 突然大量に増量する必要が生じた場合、ポンプ又はカテーテルの不具合(移動、外れ、中折れなど)が疑われるので、ポンプ内の薬液残量検査、X線検査等により確認すること。また、耐薬性発現との判別を行うこと。[15.1.2参照]
7.10 用量の調整には、痙縮が循環器系機能の維持及び深部静脈血栓症を予防している可能性のあることも考慮し、立位、歩行のバランス維持など日常生活動作を適切に保持するために、ある程度の痙縮を残すことも検討すること。
7.11 用量設定期及び維持期において使用が推奨される製剤(1日用量別)は次のとおり。
| 1日用量 | 使用が推奨される製剤 |
| 200μg未満 | 髄注0.05% |
| 200μg以上、300μg未満 | 髄注0.05%又は髄注0.2% |
| 300μg以上、600μg以下 | 髄注0.2% |
8. 重要な基本的注意
8.1 離脱症状
本剤の長期連用中に投与が突然中止・中断されると、高熱、精神状態の変化(幻覚、錯乱、興奮状態等)、痙攣発作、リバウンド症状としての痙縮の増強、筋硬直などの症状が発現し、まれに横紋筋融解症、多臓器不全及び死に至ることもあるとの報告があるので、投与を中止する場合は、用量を徐々に減量するなど慎重に行うこと。海外の市販後12年間の調査で82例(死亡に至った17例を含む)の離脱症状が報告されている。通常、離脱症状は本剤の投与中止・中断後数時間から数日以内に発現している。また、離脱症状の臨床的特徴は、自律神経反射異常、感染症(敗血症)、悪性高体温症、神経遮断性悪性症候群、あるいは代謝亢進状態や広範な横紋筋融解症等に類似することもあるので鑑別に注意すること。[1.2、7.3参照]
8.1.1 一般的な原因
本剤における離脱症状は、カテーテルのトラブル(特に外れ)、ポンプ内の薬液不足、ポンプの電池切れ、又は誤った用量設定等が原因で、発現するおそれがある。ポンプ、カテーテル及びプログラマ(専用の用量調整用の体外プログラミング機器)の説明書を熟読の上、ポンプシステムのプログラミング及びモニタリング、薬液の補充スケジュール及びその手順、ならびにポンプのアラームに十分注意すること。患者及び介護者には薬液補充のための受診の重要性及び離脱症状の初期症状(投与により改善していた痙縮の増悪、そう痒症、血圧低下及び感覚異常)について十分説明し、異常がみられた場合には直ちに受診するよう指導すること。
8.1.2 処置
離脱症状に対する治療として、投与中止・中断前の用量あるいはそれに近い用量での本剤の投与再開が推奨される。投与再開が遅れる場合は、バクロフェンの経口投与、ベンゾジアゼピン系薬剤(ジアゼパム等)の経口、経腸、又は静脈内投与により症状の重篤化を予防できることがある。
8.2 眠気等を催すことがあるので、本剤投与中の患者には自動車の運転等危険を伴う機械の操作には従事させないよう注意すること。
8.3 本剤の投与に際しては、離脱症状、過量投与等による副作用が発現するおそれがあり、患者又はそれに代わり得る適切な者に対して、これらの初期症状について十分に説明し、異常を感じた場合には、直ちに医師に連絡し、指示を仰ぐよう注意を与えること。
8.4 本剤による治療は、原因療法ではなく対症療法であることに留意し、リハビリテーション等の導入について十分に考慮すること。
8.5 海外において感染による髄膜炎が報告されているため、本剤の投与に際しては、投与部位からの感染に十分注意し、異常が認められた場合には髄液検査を実施するなど、適切な処置を行うこと。また、髄液漏による頭痛が発現することがあるので、髄液漏に十分注意し、発現が認められた場合には適切な処置を行うこと。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 てんかん患者及びその既往歴のある患者
症状を誘発するおそれがある。[9.7.2参照]
9.1.2 精神障害のある患者
精神症状が悪化するおそれがある。
9.1.3 消化性潰瘍のある患者
腹痛等の消化器系の副作用が報告されており、症状が悪化するおそれがある。
9.1.4 呼吸不全のある患者
本剤の筋弛緩作用により呼吸抑制があらわれるおそれがある。
9.1.5 自律神経反射異常の既往歴を有する患者
侵害受容刺激あるいは本剤の突然の中止により、自律神経系反射異常発作が起こるおそれがある。
9.1.6 低体重の患者
安全性は確立していない(使用経験が少ない)。
9.1.7 感染症を有する患者
効果判定が妨げられる場合があるため、スクリーニング時に感染症に罹患していないことを確認すること。また、感染症により手術に伴う合併症のリスクが高まるため、ポンプシステム植込み前にも患者が感染症に罹患していないことを確認すること。ポンプシステム植込み後に感染症に罹患した場合には、用量調整が困難になることがあるので注意すること。
9.1.8 髄液の循環異常を示す患者
本剤の循環が正常でないため本剤の作用が変化する可能性がある。
9.2 腎機能障害患者
用量の調節に注意すること。本剤は大部分が未変化体のまま尿中に排泄されるため、血中濃度が上昇するおそれがある。
9.3 肝機能障害患者
症状が悪化するおそれがある。
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。動物実験(妊娠ラット静脈内投与試験)で胎盤を通過することが報告されている。
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。動物実験(分娩後ラット静脈内投与試験)で乳汁中に移行することが報告されている。
9.7 小児等
9.7.1 ポンプ植込みに十分な体格であることを考慮すること(本剤専用のポンプの電子添文を参照すること)。
9.7.2 特にてんかん及びその既往歴のある患者では発作を誘発するおそれがある。[9.1.1参照]
9.7.3 3歳未満の患者における長期持続投与による使用経験は得られていない。
9.8 高齢者
低用量(25μg)から投与を開始するなど患者の状態を観察しながら慎重に投与すること。生理機能が低下していることが多く、比較的低用量で筋力低下、倦怠感等があらわれることがある。
10. 相互作用
10.2 併用注意
| 降圧薬 | 降圧作用を増強するおそれがある。 | 相互に作用を増強すると考えられている。 |
| 中枢神経抑制薬 催眠鎮静薬、抗不安薬、麻酔薬等 アルコール | 中枢神経抑制作用を増強するおそれがある。 | 相互に作用を増強すると考えられている。 |
| オピオイド系鎮痛剤 モルヒネ等 | 低血圧あるいは呼吸困難等の副作用を増強するおそれがある。 | 相互に作用を増強すると考えられている。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。なお、副作用の発現頻度は、全例調査方式で行った国内使用成績調査の結果に基づき算出した。
11.1.1 依存性(頻度不明)
バクロフェンの経口投与により幻覚・錯乱等が発現したという報告があり、精神依存形成につながるおそれがある。
11.2 その他の副作用
| 0.1〜3%未満 | 0.1%未満 | 頻度不明 | |
| 精神神経系 | 頭痛、傾眠、痙攣発作、筋緊張低下、しびれ、嗜眠、昏睡、歩行困難、筋緊張(亢進) | 幻覚、情緒不安定、うつ状態、会話障害 | 感覚減退、錯感覚、見当識障害、思考異常、アジテーション、重圧感、不眠症、言語機能障害、反応性遅延、無力症、頸部痛、背部痛、振戦、視神経調節障害 |
| 循環器 | 血圧低下 | 徐脈 | 期外収縮、高血圧 |
| 呼吸器 | 肺炎 | 呼吸困難、低換気 | 鼻咽頭炎、呼吸抑制 |
| 消化器 | 悪心、嘔気(嘔吐)、腹部膨満感、便秘、下痢(便失禁) | 胃部不快感、排便障害 | 口内乾燥、唾液分泌亢進 |
| 泌尿器・生殖器 | 排尿困難、尿失禁、尿閉 | 性機能障害 | 頻尿、副睾丸炎、前立腺炎、前立腺特異性抗原増加 |
| 過敏症 | 発疹等 | そう痒症 | |
| 全身症状 | 発熱、脱力感、異常感、めまい(ふらつき)、疼痛、筋力低下 | 悪寒、倦怠感 | ほてり、灼熱感 |
| その他 | 冷感、CK上昇、胸部不快感 | 浮腫、耳管開放、皮膚潰瘍、転倒、CRP上昇、LDH上昇、カテーテル留置部位異常感覚、四肢重感 |
13. 過量投与
カテーテルの開存性又は位置を確認する際、カテーテル内の薬液を不注意に送達することにより過量投与が生じることがある。また、ポンプシステムのプログラミングミス、極端に急激な増量、経口バクロフェンとの併用、あるいはポンプの機能異常等が原因で発現することがある。[7.3参照]
13.1 症状
特徴的な症状は傾眠、意識障害、呼吸抑制、昏睡等の中枢神経抑制症状である。また、痙攣、錯乱、幻覚、全身筋緊張低下、反射低下・消失、血圧低下、徐脈、低体温等があらわれることがある。
13.2 処置
速やかにポンプを停止させる(プログラマが無い場合には、ポンプ内の残存薬液をすべて抜き取ることでも薬液注入は停止する)。呼吸抑制がみられる場合、人工呼吸あるいは必要に応じて挿管するとともに心血管系の機能保持のための処置を行う。本剤は主として腎から排泄されるため、水分の供給を十分に行い、可能ならば利尿薬を併用する。腎機能が低下している場合には血液透析等を考慮する。痙攣が発現した場合にはジアゼパムを慎重に静脈内注射する。症状の発現直後であれば、髄液中バクロフェン濃度を低下させるために、腰椎穿刺又はポンプアクセスポートより30〜40mLの髄液を抜き取ることも有効である。ただし、その場合、低髄圧症状、ヘルニア等の発現に注意しながら急激には抜き取らないこと。なお、過量投与による症状が改善した後もポンプを停止させたままで放置した場合には、離脱症状が発現する可能性があるため、症状が改善した後には、患者の痙縮の状態を十分観察しながら、過量投与を起こす前の用量あるいはそれに近い用量で本剤の投与を再開すること。
14. 適用上の注意
14.1 薬剤投与時の注意
14.1.1 本剤の長期持続投与は、本剤の髄腔内持続投与用に承認された専用の植込み型プログラマブルポンプを用いること。本ポンプは本剤を保存するリザーバを内蔵し、本剤の充填は、注射器に0.22μmのフィルターを必ず装着し、ポンプの充填用薬剤注入口へ行う。本ポンプは、体外からの専用プログラマを使用して用量の変更が可能である。本ポンプはいくつかの投与モードを内蔵しているが、臨床試験は主に単純連続モードで実施されており、単純連続モード以外のモードに関する有効性及び安全性は確立されていない。詳細に関しては、本ポンプの電子添文、説明書等を参照すること。
14.1.2 離脱症状や過量投与は、一般にカテーテル及びポンプの障害、誤った用量設定等によって起こるおそれがあるので、ポンプ、カテーテル及びプログラマ等の説明書の指示及び注意に従い、ポンプ及びカテーテルの植込み、本剤の補充、用量の調節等を適切に行うこと。
14.1.3 本剤は、いずれのアンプルも1回使い切りの製剤であり、未使用の残液は廃棄すること。
14.1.4 薬液を補充する際は、ポンプ内の薬液を抜き取り、新しい薬液を補充すること。また、薬液の補充は、前回の充填から3ヵ月以内に行うこと。
14.1.5 本剤のスクリーニングならびにポンプシステム植込み時に、頭痛、悪心、嘔吐等を発現することがある。意思表示をできない小児等の場合、観察を十分に行い、必要に応じて適切な処置を行うこと。
15. その他の注意
15.1 臨床使用に基づく情報
15.1.1 本剤は錐体外路系疾患(パーキンソン症候群、アテトーシス等)の治療には適当でない。
15.1.2 本剤投与中に本剤に対し耐薬性を生じ、効果が減弱することがある。米国の臨床試験では4.1%(27/662例)に耐薬性が認められ、本剤の休薬が行われている。ポンプ又はカテーテルの不具合(移動、外れ、中折れなど)によって、効果が減弱する場合もあるので、ポンプ内の薬液残量検査、X線検査等によりポンプ又はカテーテルに不具合がないか確認すること。耐薬性が発現したと判断された場合には、本剤の投与を2〜4週間休止する。休止にあたっては、本剤の急激な投与中断による離脱症状の発現に注意し、投与量を徐々に減量するなど慎重に行うこと。なお、本剤の投与再開は、用量設定期における初回投与量から始めること。[7.9参照]
16. 薬物動態
16.1 血中濃度
重度痙性麻痺患者8例にバクロフェン50μgを単回髄腔内投与したとき、投与2時間後までの髄液中濃度は350〜1320ng/mLであり、2〜4時間後では29〜950ng/mLであった。投与1〜4時間後までの血漿中濃度は0.4〜0.6ng/mLであった(バクロフェン内服剤の併用4例を除く)1)。
雄性イヌに14C-バクロフェン250μg(約0.019mg/kg)を単回髄腔内投与したとき、髄液中放射能濃度は投与0.58時間後でCmax(11263.7ng/mL)に達し、生物学的半減期は0.54時間(投与2又は4時間後まで)及び8.2時間(投与4又は6時間後以降)であった。血漿中放射能濃度は投与0.50時間後でCmax(20.5ng/mL)に達し、生物学的半減期は8.0時間であった2)。
16.4 代謝
バクロフェンを経口投与したとき、大部分が未変化体として存在するが、一部は酸化的脱アミノ化されて4-hydroxy-3-(4-chlorophenyl)butyric acidに代謝された3)。
16.5 排泄
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国内第III相臨床試験
重度痙性麻痺患者30例[脊髄損傷12例、脊髄小脳変性症4例、脊髄血管障害3例、後縦靭帯骨化症1例、頸部脊椎症1例、脳性麻痺2例、頭部外傷2例及び小児脳性麻痺(7歳以上)5例]を対象とした、本剤25μg(4例:成人1例、小児3例)、50μg(25例:成人23例、小児2例)又は75μg(成人1例)の単回髄腔内投与によるスクリーニング試験の結果、主要評価項目である下肢平均Ashworth評点は投与前3.79から投与4時間後には1.76となり、有意な低下が認められた。「有効」例数の割合は96.7%(29/30例)であった。さらに、スクリーニング試験における有効例を対象とした本剤専用ポンプシステム植込み後6ヵ月までの長期持続投与試験(25例:成人20例、小児5例)ならびに6ヵ月以降長期フォローを行った長期安全性試験(24例:成人19例、小児5例)の結果、主要評価項目の下肢平均Ashworth評点について成人患者では36ヵ月後まで、小児患者では27ヵ月後までの各評価時期で有意な低下が認められ、抗痙縮効果が長期間持続することが確認された6)。
ポンプシステム植込み後のAshworth評点の変化(国内)
副作用は、成人では72.0%(18/25例)に認められ、主な副作用は、頭痛28.0%(7/25例)、無力症16.0%(4/25例)、感覚減退16.0%(4/25例)であった。小児では80.0%(4/5例)に認められ、主な副作用は、CK上昇40.0%(2/5例)、LDH上昇20.0%(1/5例)、血圧低下20.0%(1/5例)であった。
17.1.2 海外臨床試験
脊髄損傷による重度痙性麻痺患者を対象としたスクリーニング試験の結果は次のとおりで、主要評価項目である下肢平均Ashworth評点の有意な低下がみられ、本剤による抗痙縮効果が確認された。また長期持続投与試験の結果、抗痙縮効果は24ヵ月以上にわたり持続することが確認された。
| 原疾患名 | 症例数 | 下肢平均Ashworth評点 | ||
| 投与前 | 4時間後 | 検定注1) | ||
| 脊髄損傷 | 31 | 3.80 | 1.56 | P<0.001 |
| 多発性硬化症 | 12 | 3.78 | 1.51 | P<0.001 |
有害事象発現頻度は、全期間を通じて66.0%(66/100例)であり、主な有害事象は、脱力感24.0%(24/100例)、筋緊張低下17.0%(17/100例)、傾眠11.0%(11/100例)であった7)。
17.1.3 海外臨床試験
脊髄損傷による重度痙性麻痺患者を対象としたスクリーニング試験の結果は次のとおりで、主要評価項目である下肢平均Ashworth評点の有意な低下がみられ、本剤による抗痙縮効果が確認された。また長期持続投与試験の結果、抗痙縮効果は24ヵ月以上にわたり持続することが確認された。
| 原疾患名 | 症例数 | 下肢平均Ashworth評点 | ||
| 投与前 | 4時間後 | 検定注1) | ||
| 脊髄損傷 | 15 | 3.8 | 1.1 | P<0.0001 |
| 多発性硬化症 | 14 | 4.2 | 1.2 | P<0.0001 |
有害事象発現頻度は、全期間を通じて94.1%(32/34例)であり、主な有害事象は、脱力感43.8%(14/34例)、傾眠34.4%(11/34例)、浮腫15.6%(5/34例)であった8)。
17.1.4 海外臨床試験
脳性麻痺(4歳以上)による重度痙性麻痺患者を対象としたスクリーニング試験の結果は次のとおりで、主要評価項目である下肢平均Ashworth評点の有意な低下がみられ、本剤による抗痙縮効果が確認された。また長期持続投与試験の結果、抗痙縮効果は24ヵ月以上にわたり持続することが確認された。
| 原疾患名注2) | 症例数 | 下肢平均Ashworth評点 | ||
| 投与前 | 4時間後 | 検定注1) | ||
| 脳性麻痺 | 51 | 3.36 | 2.14 | P<0.001 |
| (4〜6歳) | (14) | (3.29) | (2.14) | (P<0.001) |
| (7〜16歳) | (29) | (3.48) | (2.14) | (P<0.001) |
| (17歳〜) | (8) | (3.36) | (2.45) | (P=0.02) |
有害事象発現頻度は、全期間を通じて82.4%(42/51例)であり、主な有害事象は、筋緊張低下31.4%(16/51例)、傾眠25.5%(13/51例)、痙攣発作23.5%(12/51例)であった9)。
17.1.5 海外臨床試験
脳性麻痺(4歳以上)による重度痙性麻痺患者を対象としたスクリーニング試験の結果は次のとおりで、主要評価項目である下肢平均Ashworth評点の有意な低下がみられ、本剤による抗痙縮効果が確認された。また長期持続投与試験の結果、抗痙縮効果は24ヵ月以上にわたり持続することが確認された。
| 原疾患名注2) | 症例数 | 下肢平均Ashworth評点 | ||
| 投与前 | 4時間後 | 検定注1) | ||
| 脳性麻痺 | 48 | 2.77 | 1.99 | P<0.001 |
| (4〜6歳) | (7) | (2.82) | (1.83) | (P=0.02) |
| (7〜16歳) | (26) | (2.60) | (1.85) | (P<0.01) |
| (17歳〜) | (15) | (2.99) | (2.22) | (P<0.01) |
有害事象発現頻度は、全期間を通じて66.7%(54/81例)であり、主な有害事象は、筋緊張低下29.6%(24/81例)、頭痛23.5%(19/81例)、傾眠22.2%(18/81例)であった10)。
17.1.6 海外臨床試験
外傷等の脳損傷による重度痙性麻痺患者を対象としたスクリーニング試験の結果は次のとおりで、主要評価項目である下肢平均Ashworth評点の有意な低下がみられ、本剤による抗痙縮効果が確認された。また長期持続投与試験の結果、抗痙縮効果は24ヵ月以上にわたり持続することが確認された。
| 原疾患名注2) | 症例数 | 下肢平均Ashworth評点 | ||
| 投与前 | 4時間後 | 検定注1) | ||
| 外傷等の脳損傷 | 11 | 4.16 | 2.16 | P=0.001 |
有害事象は、全期間を通じて発現しなかった11)。
注1)Wilcoxon符号付順位和検定(投与前値と4時間後値の比較)
注2)50μg投与の成績
17.2 製造販売後調査等
17.2.1 国内使用成績調査
脳脊髄疾患由来の重度の痙性麻痺患者に対して、使用実態下における単回髄腔内投与によるスクリーニング及びシンクロメッドポンプを用いた長期持続投与の有効性及び安全性を検討することを目的とした中央登録方式による全例調査を実施し、国内218施設から1,478例の症例を収集した。スクリーニング期及び長期持続投与期の有効性解析対象症例は各々1,472例、742例であった。なお、原疾患の内訳は、スクリーニング期で脊髄損傷262例、脊髄血管障害48例、後縦靭帯骨化症24例、頸部脊椎症39例、多発性硬化症29例、脊髄小脳変性症223例、痙性脳性麻痺197例、頭部外傷94例等であり、長期持続投与期で脊髄損傷136例、脊髄血管障害22例、後縦靭帯骨化症10例、頸部脊椎症14例、多発性硬化症18例、脊髄小脳変性症105例、痙性脳性麻痺107例、頭部外傷52例等であった。
スクリーニング期の下肢平均Ashworth評点は、投与前2.95から投与後1.79となり、有意な低下が認められた。また、上肢平均Ashworth評点は、投与前2.50から投与後1.96となり、有意な低下が認められた。スクリーニング期における「有効」例数の割合は、87.6%(1,290/1,472例)であった。
ポンプシステム植込み後12ヵ月間フォローを行った長期持続投与期の下肢平均及び上肢平均Ashworth評点は、いずれも各評価時期で有意な低下が認められ、抗痙縮効果が長期間持続することが確認された12)。
スクリーニング期の下肢平均Ashworth評点は、投与前2.95から投与後1.79となり、有意な低下が認められた。また、上肢平均Ashworth評点は、投与前2.50から投与後1.96となり、有意な低下が認められた。スクリーニング期における「有効」例数の割合は、87.6%(1,290/1,472例)であった。
ポンプシステム植込み後12ヵ月間フォローを行った長期持続投与期の下肢平均及び上肢平均Ashworth評点は、いずれも各評価時期で有意な低下が認められ、抗痙縮効果が長期間持続することが確認された12)。
| 時点 | 下肢平均Ashworth評点 | 上肢平均Ashworth評点 | ||||
| 例数 | 平均値±標準偏差 | 検定注3) | 例数 | 平均値±標準偏差 | 検定注3) | |
| 投与前 | 724 | 3.11±0.91 | − | 470 | 2.59±0.94 | − |
| 1ヵ月後 | 701 | 1.97±0.82 | P<0.0001 | 450 | 1.95±0.81 | P<0.0001 |
| 3ヵ月後 | 660 | 1.93±0.79 | P<0.0001 | 427 | 1.89±0.78 | P<0.0001 |
| 6ヵ月後 | 646 | 1.94±0.82 | P<0.0001 | 414 | 1.90±0.79 | P<0.0001 |
| 12ヵ月後 | 629 | 1.91±0.82 | P<0.0001 | 401 | 1.88±0.81 | P<0.0001 |
18. 薬効薬理
18.1 作用機序
バクロフェンはγ-アミノ酪酸(GABA)の誘導体で、脊髄の単シナプス及び多シナプス反射の両方を抑制し、γ-運動ニューロンの活性を低下させる抗痙縮剤である。
18.2 脊髄反射の抑制作用
18.3 運動ニューロン活性の抑制作用
18.4 実験的固縮の抑制作用
18.5 筋電図学的改善作用
18.6 鎮痛作用
19. 有効成分に関する理化学的知見
19.1. バクロフェン
| 一般的名称 | バクロフェン |
|---|---|
| 一般的名称(欧名) | Baclofen |
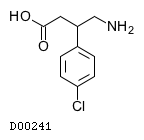
| 化学名 | (3RS)-4-Amino-3-(4-chlorophenyl)butanoic acid |
| 分子式 | C10H12ClNO2 |
| 分子量 | 213.66 |
| 物理化学的性状 | 白色〜微黄白色の結晶性の粉末である。酢酸(100)に溶けやすく、水に溶けにくく、メタノール又はエタノール(95)に極めて溶けにくく、ジエチルエーテルにほとんど溶けない。希塩酸に溶ける。 |
| KEGG DRUG |
|
21. 承認条件
本剤の安全性及び有効性を十分に理解し、施術に関する十分な知識・経験のある医師によってのみ用いられるよう、本剤を納入する前に予め講習(ポンプシステムに関する事項を含む)を実施する等の適切な措置を講じること。
22. 包装
<ギャバロン髄注0.005%>
1mL 1アンプル
<ギャバロン髄注0.05%>
20mL 1アンプル
<ギャバロン髄注0.2%>
5mL 1アンプル
23. 主要文献
- 社内資料:国内第III相試験(DL4040-01試験)(2005年4月11日承認、CTD2.7.2.2)
- 社内資料:薬物動態試験(2005年4月11日承認、CTD2.6.4.4)
- Faigle JW,et al., Postgrad Med J., 48 (S-5), 9-13, (1972)
- 高杉紀雄ほか, 日本薬学会講演要旨集(第97回), 237, (1977)
- 社内資料:薬物動態試験(2005年4月11日承認、CTD2.6.4.6)
- 社内資料:国内臨床成績(2007年1月26日承認、CTD2.7.6)
- 社内資料:海外臨床成績(2005年4月11日承認、CTD2.7.6.5)
- 社内資料:海外臨床成績(2005年4月11日承認、CTD2.7.6.4)
- Gilmartin R,et al., J Child Neurol., 15 (2), 71-77, (2000) »PubMed »DOI
- 社内資料:海外臨床成績(2005年4月11日承認、CTD2.7.6.7)
- Meythaler JM,et al., Arch Phys Med Rehabil., 77 (5), 461-466, (1996) »PubMed »DOI
- 山口広貴ほか, 臨床医薬, 32 (6), 419-467, (2016)
- Schwarz M,et al., Local-spinal therapy of spasticity., 65-79, (1988), (Berlin:Springer-Verlag)
- Kroin JS,et al., Exp Brain Res., 54 (1), 191-194, (1984) »PubMed
- 福田英臣ほか, 応用薬理, 13 (5), 611-626, (1977)
- Fehr HU,et al., J Int Med Res., 2, 36-47, (1974)
- Kuroiwa M,et al., Pharmacol Res., 60 (5), 392-396, (2009) »PubMed
- 津山直一ほか, 薬理と治療, 4 (4), 959-970, (1976)
- 糸賀叡子ほか, 診断と治療, 64 (9), 1772-1776, (1976)
- 玄番央恵ほか, 臨床脳波, 19 (6), 395-399, (1977)
- Wilson PR,et al., Eur J Pharmacol., 51 (4), 323-330, (1978) »PubMed
- Yaksh TL,et al., Anesthesiology, 54 (6), 451-467, (1981) »PubMed
24. 文献請求先及び問い合わせ先
文献請求先
第一三共株式会社
製品情報センター
〒103-8426
東京都中央区日本橋本町3-5-1
電話:0120-189-132
製品情報問い合わせ先
第一三共株式会社
製品情報センター
〒103-8426
東京都中央区日本橋本町3-5-1
電話:0120-189-132
26. 製造販売業者等
26.1 製造販売元
第一三共株式会社
東京都中央区日本橋本町3-5-1
26.2 提携先
メドトロニック社
米国