医薬品情報
| 総称名 | サーティカン |
|---|---|
| 一般名 | エベロリムス |
| 欧文一般名 | Everolimus |
| 製剤名 | エベロリムス錠 |
| 薬効分類名 | 免疫抑制剤(mTOR阻害剤) |
| 薬効分類番号 | 3999 |
| ATCコード | L04AH02 |
| KEGG DRUG |
D02714
エベロリムス
|
| KEGG DGROUP |
DG01597
mTOR阻害薬
|
| JAPIC | 添付文書(PDF) |
添付文書情報2019年7月 改訂(第1版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| サーティカン錠0.25mg | Certican Tablets | ノバルティスファーマ | 3999022F1028 | 225.3円/錠 | 劇薬, 処方箋医薬品 |
| サーティカン錠0.5mg | Certican Tablets | ノバルティスファーマ | 3999022F2024 | 456.9円/錠 | 劇薬, 処方箋医薬品 |
| サーティカン錠0.75mg | Certican Tablets | ノバルティスファーマ | 3999022F3020 | 651.8円/錠 | 劇薬, 処方箋医薬品 |
1. 警告
1.1 心移植、腎移植、肝移植における本剤の投与は、免疫抑制療法及び移植患者の管理に精通している医師又はその指導のもとで行うこと。
2. 禁忌
次の患者には投与しないこと
4. 効能または効果
下記の臓器移植における拒絶反応の抑制
心移植、腎移植、肝移植
6. 用法及び用量
<心移植>
通常、成人にはエベロリムスとして1.5mgを、1日2回に分けて経口投与する。なお、開始用量は1日量として3mgまでを用いることができる。患者の状態やトラフ濃度によって適宜増減する。
<腎移植>
通常、成人にはエベロリムスとして1.5mgを、1日2回に分けて経口投与する。患者の状態やトラフ濃度によって適宜増減する。
<肝移植>
通常、成人にはエベロリムスとして2.0mgを、1日2回に分けて経口投与する。患者の状態やトラフ濃度によって適宜増減する。なお、原則、エベロリムスの投与開始は移植後4週以降とする。
7. 用法及び用量に関連する注意
<効能共通>
7.1 食事の影響があるため、食後又は空腹時のいずれかの一定の条件下で投与し、本剤の血中トラフ濃度を測定し、投与量を調節すること。[16.2.1参照]
7.2 カルシニューリン阻害薬及び副腎皮質ホルモン剤と併用すること。カルシニューリン阻害薬を併用しない場合、十分な効果が得られないおそれがある。本剤の類薬(シロリムス)の試験において、移植3ヵ月後にシクロスポリンの投与を中止した腎移植患者において、急性拒絶反応の発現率がシクロスポリンの投与を継続した患者に比べて有意に増加したとの報告がある1)。また、海外臨床試験において、移植5ヵ月目にタクロリムスの投与を中止した肝移植患者において、急性拒絶反応の発現率がタクロリムスの投与を継続した患者に比べて有意に増加した2)。
7.3 本剤の全血中濃度を定期的に測定すること。[16.1.1-16.1.4、16.8.1参照]曝露量と有効性、及び曝露量と安全性の関連についての解析から、本剤の血中トラフ濃度(C0)が3.0ng/mL以上の患者では、3.0ng/mL未満の患者に比べて急性拒絶反応の発現率が低いことが認められている。推奨される本剤の治療濃度の上限は8ng/mLである。12ng/mLを超える濃度での有効性及び安全性の検討は実施されていない。
7.4 本剤の用量調節は、用量変更から4〜5日以上経過してから測定した本剤の血中トラフ濃度(C0)に基づいて行うことが望ましい。シクロスポリンは本剤のバイオアベイラビリティを増加させるため、シクロスポリンの血中濃度が大幅に低下すると(血中トラフ濃度(C0)<50ng/mL)、本剤の血中濃度が低下するおそれがある。[8.2、16.7.1参照]
7.5 肝機能障害を有する患者では、本剤の血中トラフ濃度(C0)を頻繁に測定すること。
軽度又は中等度の肝機能障害(Child-Pugh分類クラスA又はB)を有する患者が以下の3項目の内2項目以上に該当する場合には、用量を通常量の約半量に減量すること:ビリルビン>2mg/dL、アルブミン<3.5g/dL、プロトロンビン時間>1.3INR(4秒を超える延長)。
更に、本剤の血中濃度に基づいて用量調節を行うこと。[9.3、16.6.2参照]
軽度又は中等度の肝機能障害(Child-Pugh分類クラスA又はB)を有する患者が以下の3項目の内2項目以上に該当する場合には、用量を通常量の約半量に減量すること:ビリルビン>2mg/dL、アルブミン<3.5g/dL、プロトロンビン時間>1.3INR(4秒を超える延長)。
更に、本剤の血中濃度に基づいて用量調節を行うこと。[9.3、16.6.2参照]
7.6 本剤は併用するシクロスポリンの腎毒性を増強するおそれがある。また、本剤とシクロスポリン又はタクロリムスの併用により腎障害が発現するおそれがあるため、腎移植患者、肝移植患者及び維持期の心移植患者ではシクロスポリン又はタクロリムスの用量を減量すること。なお、シクロスポリン又はタクロリムスの用量は、シクロスポリン又はタクロリムスの血中トラフ濃度(C0)に基づいて調節する。[[8.5、9.2、11.1.1、17.1.1-17.1.5参照]表「シクロスポリンの血中トラフ濃度(C0)の記述統計量(B253試験、A1202試験、A2309試験)」、「タクロリムスの血中トラフ濃度(C0)の記述統計量(H2307試験、H2304試験)」参照]
7.7 シクロスポリンとの併用にあたってはシクロスポリンのマイクロエマルジョン製剤と同時投与が望ましい。
7.8 本剤と併用するシクロスポリン又はタクロリムスを減量する前に、本剤の定常状態の血中トラフ濃度(C0)が3ng/mL以上であることを確認すること。
<心移植>
7.9 心移植における本剤の用量設定の際には、下記を参照すること。(心移植患者を対象として、標準量のシクロスポリンのマイクロエマルジョン製剤及び副腎皮質ホルモン剤と併用した本剤1.5mg/日及び3mg/日の有効性及び安全性をアザチオプリン1〜3mg/kg/日と比較した海外第III相試験(B253試験)の結果)
7.9.1 本剤(1.5mg/日及び3mg/日)の平均血中トラフ濃度別の有効性及び副作用発現率
| 本剤の平均血中トラフ濃度(ng/mL) | グレード3A(ISHLT)以上の急性拒絶反応発現率 | 副作用発現率 |
| 3未満 | 44.1%(30/68) | 64.4%(47/73) |
| 3〜4未満 | 32.7%(16/49) | 63.0%(34/54) |
| 4〜5未満 | 18.6%(8/43) | 62.5%(25/40) |
| 5〜6未満 | 22.0%(11/50) | 57.5%(23/40) |
| 6〜7未満 | 18.9%(7/37) | 53.3%(16/30) |
| 7〜8未満 | 23.8%(10/42) | 60.0%(18/30) |
| 8〜9未満 | 21.4%(6/28) | 63.0%(17/27) |
| 9〜10未満 | 15.0%(3/20) | 60.9%(14/23) |
| 10以上 | 16.4%(11/67) | 77.2%(44/57) |
| 本剤の平均血中トラフ濃度の確認できた全症例 | − | 63.6%(238/374) |
| 本剤投与全症例 | 26.4%(111/420) | 66.2%(278/420) |
7.9.2 移植後1年間の時期別副作用発現率
| 移植後経過期間 | 本剤1.5mg/日投与 | 本剤3mg/日投与 |
| 〜5日 | 15.8%(33/209) | 13.7%(29/211) |
| 6日〜14日(2週) | 9.3%(19/204) | 13.5%(28/207) |
| 15日〜30日(1ヵ月) | 23.1%(46/199) | 30.7%(62/202) |
| 31日〜90日(3ヵ月) | 23.0%(44/191) | 36.1%(69/191) |
| 91日〜365日(1年) | 40.1%(73/182) | 49.1%(84/171) |
7.9.3 本剤の血中トラフ濃度の経時推移
| 本剤の投与期間 | 本剤1.5mg/日投与 | 本剤3mg/日投与 | ||
| 血中トラフ濃度(ng/mL) | 例数 | 血中トラフ濃度(ng/mL) | 例数 | |
| 2日目 | 1.8±2.7 | 148 | 4.2±3.6 | 157 |
| 1週目 | 5.4±3.7 | 159 | 10.2±6.8 | 159 |
| 2週目 | 5.4±4.0 | 159 | 10.0±7.2 | 173 |
| 3週目 | 5.2±4.4 | 155 | 10.2±6.6 | 150 |
| 1ヵ月目 | 5.4±3.9 | 147 | 8.9±6.0 | 135 |
| 2ヵ月目 | 5.1±3.5 | 152 | 8.7±5.1 | 141 |
| 3ヵ月目 | 5.1±3.8 | 143 | 9.1±6.3 | 133 |
| 6ヵ月目 | 4.8±3.3 | 108 | 8.5±5.6 | 109 |
8. 重要な基本的注意
8.1 シクロスポリン、タクロリムス及び副腎皮質ホルモン剤との併用に際しては、各薬剤の添付文書に記載されている「警告」、「禁忌」、「併用禁忌」、「重要な基本的注意」、「特定の背景を有する患者に関する注意」、「重大な副作用」等の使用上の注意を必ず確認すること。
8.2 シクロスポリンの併用により本剤のバイオアベイラビリティは有意に増加する。健康成人を対象とした単回投与試験において、本剤にシクロスポリンのマイクロエマルジョン製剤を併用投与したところ、単独投与時に比べて本剤のAUCは168%(範囲46%〜365%)、Cmaxは82%(範囲25%〜158%)増加した。従って、シクロスポリンの用量を変更する場合には、本剤の用量調節が必要であると考えられる。[7.4、16.7.1参照]なお、シクロスポリンのマイクロエマルジョン製剤を投与中の心移植患者において、シクロスポリンの薬物動態に対する本剤の臨床的影響はごく軽微であった。
8.3 ダイレクトクロスマッチ陽性等、抗ドナー抗体等の拒絶反応のリスク因子を有する患者を対象とした適切な臨床試験は実施されていない。
8.4 定期的に血清脂質の検査を行うこと。高脂血症がみられた場合には、適切な食事指導を実施し、必要により高脂血症用剤を投与するなど適切な処置を行うこと。[9.1.3参照]
8.6 汎血球減少、白血球減少、貧血、血小板減少、好中球減少があらわれることがあるので定期的に血液検査(血球数算定等)を実施すること。[11.1.7参照]
8.7 特に心移植患者において、心嚢液貯留があらわれることがあるので、使用に際しては心電図、心エコー、胸部X線検査等を行うこと。[11.1.13参照]
8.8 高血糖の発現、糖尿病の発症又は増悪することがあるので、定期的に空腹時血糖値の測定等を行うこと。[11.1.14参照]
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 感染症を合併している患者
免疫抑制により感染症が悪化するおそれがある。[11.1.2参照]
9.1.2 肝炎ウイルスキャリアの患者
肝炎ウイルスキャリアの患者に本剤を投与する場合は、肝機能検査値や肝炎ウイルスマーカーのモニタリングを行うなど、B型肝炎ウイルスの再活性化やC型肝炎の悪化の徴候や症状の発現に注意すること。免疫抑制剤を投与されたB型肝炎ウイルスキャリアの患者において、B型肝炎ウイルスの再活性化による肝炎があらわれることがある。また、HBs抗原陰性の患者において、免疫抑制剤の投与開始後にB型肝炎ウイルスの再活性化による肝炎を発症した症例が報告されている。また、C型肝炎ウイルスキャリアの患者において、免疫抑制剤の投与開始後にC型肝炎の悪化がみられることがある。[11.1.2参照]
9.1.3 高脂血症を合併している患者
治療上の有益性が、危険性を上回ると判断される場合にのみ投与すること。症状が悪化するおそれがある。[8.4参照]
9.2 腎機能障害患者
9.3 肝機能障害患者
9.5 妊婦
妊婦又は妊娠している可能性のある女性には投与しないこと。動物実験(ラット及びウサギ)で胚・胎児毒性を含む生殖発生毒性が認められたとの報告がある。
9.6 授乳婦
授乳しないことが望ましい。動物実験(ラット)において乳汁中に移行することが報告されている。
9.7 小児等
小児等の心移植、腎移植及び肝移植患者を対象とした有効性及び安全性を指標とした臨床試験は実施していない。また、乳児、幼児及び小児の肝移植患者を対象とした海外臨床試験において、成人での臨床試験と比較して移植後リンパ増殖性障害や重篤な感染症、胃腸障害の発現頻度が高いことが報告されている4)。
9.8 高齢者
9.8.1 患者の状態を観察しながら慎重に投与すること。一般に生理機能(腎機能、肝機能、免疫機能等)が低下している。
9.8.2 腎移植患者を対象とした臨床試験における母集団薬物動態解析の結果、本剤の薬物動態に65〜70歳の患者(18例)と母集団(673例)との明らかな差は認められていない。[16.6.4参照]
10. 相互作用
相互作用序文
本剤は主として肝代謝酵素CYP3A4によって代謝され、腸管に存在するCYP3A4によっても代謝される。また、本剤はP糖蛋白(Pgp)の基質でもあるため、本剤経口投与後の吸収と消失は、CYP3A4又はPgpに影響を及ぼす薬剤により影響を受けると考えられる。
CYP3A4を誘導する薬剤又は阻害する薬剤を併用したり中止する場合は、必ず本剤の血中トラフ濃度(C0)をモニタリングすること。
CYP3A4を誘導する薬剤又は阻害する薬剤を併用したり中止する場合は、必ず本剤の血中トラフ濃度(C0)をモニタリングすること。
薬物代謝酵素用語
CYP3A4
薬物代謝酵素用語
P糖蛋白(Pgp)
10.1 併用禁忌
| 生ワクチン(乾燥弱毒生麻しんワクチン、乾燥弱毒生風しんワクチン、経口生ポリオワクチン、乾燥BCG等) [2.3参照] | 免疫抑制下で生ワクチンを接種すると発症するおそれがあるので併用しないこと。 | 免疫抑制下で生ワクチンを接種すると増殖し、病原性をあらわす可能性がある。 |
10.2 併用注意
| リファンピシン | 本剤の血中濃度が低下することがあるので、併用する場合には治療上の有益性が危険性を上回る場合にのみ使用すること。 | これらの薬剤の代謝酵素(CYP3A4等)誘導作用により本剤の代謝が促進されると考えられる。 |
| 抗てんかん剤 フェノバルビタール フェニトイン カルバマゼピン等 抗HIV剤 エファビレンツ ネビラピン等 | 本剤の血中濃度が低下するおそれがあるので、併用する場合には血中濃度を参考に投与量を調節すること。 | これらの薬剤の代謝酵素(CYP3A4等)誘導作用により本剤の代謝が促進されると考えられる。 |
| アゾール系抗真菌剤 イトラコナゾール ボリコナゾール フルコナゾール等 | 本剤の血中濃度が上昇することがあるので、併用する場合には治療上の有益性が危険性を上回る場合にのみ使用すること。 | 代謝酵素(CYP3A4等)の抑制又は競合により、本剤の代謝が阻害されると考えられる。 |
| マクロライド系抗生物質 エリスロマイシン クラリスロマイシン等 カルシウム拮抗剤 ベラパミル ニカルジピン ジルチアゼム等 HIVプロテアーゼ阻害剤 ネルフィナビル インジナビル ホスアンプレナビル リトナビル等 | 本剤の血中濃度が上昇するおそれがあるので、併用する場合には血中濃度を参考に投与量を調節すること。 | 代謝酵素(CYP3A4等)の抑制又は競合により、本剤の代謝が阻害されると考えられる。 |
| オムビタスビル・パリタプレビル・リトナビル | 本剤のAUCが27倍、Cmaxが4.7倍に上昇したとの報告がある。やむを得ない場合を除き併用は避けること。やむを得ず併用する場合には、本剤の血中濃度をモニタリングするなど患者の状態を慎重に観察し、副作用発現に十分注意すること。 | リトナビルのCYP3A4阻害作用により、本剤の代謝が阻害される。 |
| 不活化ワクチン 不活化インフルエンザワクチン等 | ワクチンの効果が得られないおそれがある。 | 免疫抑制作用によってワクチンに対する免疫が得られないおそれがある。 |
| セイヨウオトギリソウ(St.John's Wort,セント・ジョーンズ・ワート)含有食品 | 本剤の血中濃度が低下するおそれがあるので、本剤投与時はセイヨウオトギリソウ含有食品を摂取しないよう注意すること。 | セイヨウオトギリソウの代謝酵素誘導作用により本剤の代謝が促進されると考えられる。 |
| グレープフルーツジュース | 本剤の血中濃度が上昇するおそれがあるので、本剤服用時は飲食を避けることが望ましい。 | グレープフルーツジュースが腸管の代謝酵素を阻害することによると考えられる。 |
| シクロスポリン [7.2、7.4、7.6-7.8、8.1、8.2参照] | シクロスポリンのマイクロエマルジョン製剤との併用により、本剤のバイオアベイラビリティが有意に増加したとの報告がある。シクロスポリンのマイクロエマルジョン製剤との併用に際しては7.2、7.4、7.6-7.8及び8.1、8.2項を参照し投与すること。 | 代謝酵素(CYP3A4等)の競合により、本剤の代謝が阻害されると考えられる。 |
| 抗ヒト胸腺細胞ウサギ免疫グロブリン サイモグロブリン | 過度の免疫抑制が起こることがある。海外で実施された新規心移植患者を対象とした臨床試験において、本剤、シクロスポリン(腎移植よりも高い血中トラフ濃度)及び副腎皮質ホルモン剤を併用し、サイモグロブリン(抗ヒト胸腺細胞ウサギ免疫グロブリン)導入療法を受けた患者集団において、移植後の3ヵ月間に重大な感染症の増加がみられた。特に過剰な免疫抑制状態となりやすい移植前の入院及び心室補助循環装置を必要とする患者においてより高い死亡率との関連が認められた。 | 共に免疫抑制作用を有するため。 |
| ミダゾラム(経口剤:国内未販売) | ミダゾラムの血中濃度が上昇するおそれがある。 | 本剤がCYP3A4の基質となる薬剤の代謝を阻害し、血中濃度を上昇させる可能性がある。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 腎障害(10.6%)
11.1.2 感染症(23.1%)
11.1.3 移植腎血栓症(0.4%:腎移植患者での頻度)
腎移植患者において、腎の動脈及び静脈の血栓症のリスク増加により、多くは移植後30日以内に移植腎廃絶に至ったとの報告がある。本剤の投与に際しては、腎血流量の低下、尿量減少等異常が認められた場合には適切な処置を行うこと。
11.1.4 肝動脈血栓症(0.2%:肝移植患者での頻度)
本剤の類薬(シロリムス)の肝移植患者を対象とした海外臨床試験において、肝動脈血栓症の発現頻度がシロリムスを投与しなかった対照群に比べて高く、その多くは移植後30日以内に発現し、移植肝廃絶や死亡に至った例も報告されている。
11.1.5 悪性腫瘍(1.8%)
悪性リンパ腫、リンパ増殖性疾患、悪性腫瘍(特に皮膚)があらわれることがある。
11.1.6 創傷治癒不良
創傷治癒不良(1.3%)や創傷治癒不良による創傷感染(1.0%)、瘢痕ヘルニア(0.7%)、創離開(0.6%)等の合併症があらわれることがある。
11.1.7 汎血球減少(1.0%)、白血球減少(8.6%)、貧血(6.3%)、血小板減少(5.8%)、好中球減少(0.9%)
血小板減少が生じた結果、消化管出血等の出血に至った症例も報告されている。[8.6参照]
11.1.8 進行性多巣性白質脳症(PML)(頻度不明)
本剤の治療期間中及び治療終了後は患者の状態を十分に観察し、意識障害、認知障害、麻痺症状(片麻痺、四肢麻痺)、言語障害等の症状があらわれた場合は、MRIによる画像診断及び脳脊髄液検査を行うとともに、投与を中止し、適切な処置を行うこと。
11.1.9 BKウイルス腎症(0.1%未満)
11.1.10 血栓性微小血管障害(0.7%)
溶血性尿毒症症候群(HUS:血小板減少、溶血性貧血、腎不全を主徴とする)、血栓性血小板減少性紫斑病(TTP)様症状(血小板減少、微小血管性溶血性貧血、腎機能障害、精神症状を主徴とする)等の血栓性微小血管障害があらわれることがある。
11.1.11 間質性肺疾患(間質性肺炎、肺臓炎)(0.6%)
死亡に至った例も報告されている。
11.1.12 肺胞蛋白症(0.1%未満)
11.1.13 心嚢液貯留(9.9%:心移植患者での頻度)
特に心移植患者において、心嚢液貯留があらわれることがある。[8.7参照]
11.1.14 高血糖(1.0%)、糖尿病の発症(2.1%)又は増悪(頻度不明)[8.8参照]
11.1.15 肺塞栓症(0.1%未満)、深部静脈血栓症(0.2%)
11.1.16 急性呼吸窮迫症候群(頻度不明)
急速に進行する呼吸困難、低酸素症、両側性びまん性肺浸潤影等の胸部X線異常等が認められた場合には投与を中止し、適切な処置を行うこと。
11.2 その他の副作用
| 5%以上 | 1%〜5%未満 | 1%未満 | 頻度不明 | |
| 血液及びリンパ系障害 | − | − | − | 凝血異常、溶血 |
| 内分泌障害 | − | − | 男性性腺機能低下(テストステロン減少、黄体形成ホルモン増加、卵胞刺激ホルモン増加) | − |
| 代謝及び栄養障害 | 高脂血症(16.0%)、高コレステロール血症、高トリグリセリド血症 | 脂質異常症 | 低カリウム血症、高尿酸血症 | − |
| 血管障害 | − | 高血圧、リンパ嚢腫 | − | − |
| 呼吸器、胸郭及び縦隔障害 | − | 胸水注)、咳嗽 | 咽頭炎 | − |
| 胃腸障害 | 下痢 | 悪心、嘔吐、口内炎、口腔内潰瘍 | 腹痛、消化不良、膵炎 | − |
| 肝胆道系障害 | − | 肝機能検査値異常、肝障害 | 黄疸、肝炎 | − |
| 皮膚及び皮下組織障害 | − | ざ瘡 | 血管神経性浮腫、発疹 | 白血球破砕性血管炎 |
| 筋骨格系及び結合組織障害 | − | 関節痛 | 筋痛 | − |
| 腎及び尿路障害 | − | 血中クレアチニン増加 | − | − |
| 全身障害及び投与局所様態 | 浮腫 | 発熱 | 疼痛 | − |
| 神経系障害 | − | 振戦 | − | − |
| その他 | − | − | − | 無精子症、卵巣嚢胞 |
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
15. その他の注意
15.2 非臨床試験に基づく情報
ラットを用いた雄性授胎能試験では、0.5mg/kg以上の用量で精巣の形態に影響が認められたほか、5mg/kg用量(治療量の範囲内)で精子運動能、精子数及び血漿中テストステロン濃度が減少し、これに伴って雄の授胎能が低下した。これらの所見は休薬による回復傾向がみられた。
16. 薬物動態
16.1 血中濃度
16.1.1 単回経口投与
健康成人24例に本剤0.5〜4mgを単独で単回経口投与したとき、全血中濃度は投与後約1時間で最高濃度に達した。消失半減期は、低用量(0.5mg及び1mg)では消失相の濃度データ(定量限界以上の値)が少なかったため算出できなかったが、2mg投与群では38.5±5.8時間、4mg投与群では34.9±2.7時間であり、ほぼ同じ値を示した。薬物動態パラメータは以下のとおりであり、投与量とCmax及びAUCの関係は線形性を示した5)(日本人のデータ、分析方法:LC/MS法)。
| 本剤の薬物動態パラメータ | 本剤の投与量 | |||
| 0.5mg | 1mg | 2mg | 4mg | |
| Tmax(hr) | 0.8(0.5〜1.0) | 1.0(0.5〜1.0) | 1.0(0.5〜1.0) | 0.8(0.5〜1.5) |
| Cmax(ng/mL) | 5.2±1.6 | 9.3±2.0 | 18.3±4.8 | 33.2±6.9 |
| Cmax/Dose(ng/mL/mg) | 10.3±3.3 | 9.3±2.0 | 9.2±2.4 | 8.3±1.7 |
| AUC0-t(ng・hr/mL) | 20±14 | 47±17 | 117±49 | 186±33 |
| AUC0-t/Dose(ng・hr/mL/mg) | 40±27 | 47±17 | 59±24 | 47±8 |
16.1.2 反復経口投与
心及び腎移植患者に本剤をシクロスポリンのマイクロエマルジョン製剤とともに1日2回投与した場合の薬物動態は、4日目までに定常状態に到達し、血中濃度の蓄積比は初回投与後の曝露量の2〜3倍であり、Tmaxは投与1〜2時間後に得られた。なお、心移植患者に本剤0.75mg及び1.5mgを投与した時の投与2、3及び6ヵ月目の本剤の定常状態薬物動態パラメータは以下のとおりであった。観察した期間を通して、Cmaxssはそれぞれの投与量で約10及び約20ng/mL、AUCτssは約80及び約160ng・hr/mL、またPTF(ピーク−トラフ濃度変動)は約80%と安定していた6)7)8)(外国人のデータ、分析方法:ELISA法)。
| 本剤の薬物動態パラメータ | 本剤0.75mg/回、1日2回投与 | 本剤1.5mg/回、1日2回投与 | ||||
| 2ヵ月目 | 3ヵ月目 | 6ヵ月目 | 2ヵ月目 | 3ヵ月目 | 6ヵ月目 | |
| 例数 | 22 | 23 | 20 | 20 | 20 | 14 |
| Cminss(ng/mL) | 4.7±2.6 | 4.9±3.0 | 4.5±2.4 | 10.0±4.3 | 10.2±5.2 | 9.8±5.6 |
| Tmax(hr) | 2(1〜5) | 2(1〜5) | 2(1〜5) | 2(1〜5) | 2(0〜5) | 2(1〜5) |
| Cmaxss(ng/mL) | 10.2±3.8 | 9.9±4.3 | 10.5±4.8 | 19.9±8.6 | 18.6±6.8 | 21.8±12.4 |
| AUCτss(ng・hr/mL) | 79±30 | 82±43 | 80±39 | 159±63 | 158±60 | 164±87 |
| Cavgss(ng/mL) | 6.6±2.5 | 6.9±3.6 | 6.7±3.3 | 13.3±5.3 | 13.1±5.0 | 13.7±7.2 |
| PTF(%) | 89±36 | 77±40 | 96±67 | 77±35 | 70±40 | 85±32 |
16.1.3 反復経口投与
(1)新規腎移植患者にシクロスポリンのマイクロエマルジョン製剤(CsA)とともに本剤1.5mg/日を開始用量として1日2回投与し、本剤の平均血中トラフ濃度(C0)を3〜8ng/mLに維持するように投与量を調節したときの血中トラフ濃度は以下のとおりであった9)(日本人のデータ、分析方法:LC/MS/MS法)。
| 評価時点(移植後) | 本剤の血中トラフ濃度 平均値±標準偏差 |
| 3日 | 3.442±1.2880(n=60) |
| 7日 | 4.711±1.4692(n=60) |
| 14日 | 5.113±1.2745(n=57) |
| 1ヵ月 | 5.155±1.3885(n=57) |
| 2ヵ月 | 5.450±1.8292(n=57) |
| 3ヵ月 | 5.349±1.4998(n=55) |
| 4ヵ月 | 5.380±1.2988(n=55) |
| 6ヵ月 | 5.497±1.5206(n=55) |
| 7ヵ月 | 5.295±1.7832(n=55) |
| 9ヵ月 | 4.897±1.1407(n=54) |
| 12ヵ月 | 5.050±1.3027(n=53) |
(2)新規腎移植患者にシクロスポリンのマイクロエマルジョン製剤とともに本剤1.5mg/日を開始用量として1日2回投与した時の投与1ヵ月目の定常状態薬物動態パラメータは以下のとおりで、Cmaxは約14ng/mL、AUCτは約90ng・hr/mL、またPTF(ピーク−トラフ濃度変動)は約120%であった9)(日本人のデータ、分析方法:LC/MS/MS法)。
| 本剤の薬物動態パラメータ | 本剤0.75mg/回、1日2回投与 |
| 1ヵ月目 | |
| 例数 | 11 |
| Cminss(ng/mL) | 4.31±1.25 |
| Tmax(hr) | 2(1〜2) |
| Cmaxss(ng/mL) | 13.5±3.5 |
| AUCτss(ng・hr/mL) | 90.7±17.7 |
| Cavgss(ng/mL) | 7.56±1.47 |
| PTF(%) | 123±32 |
16.1.4 反復経口投与
(1)新規肝移植患者にタクロリムスとともに移植後約4週から本剤2mg/日を開始用量として1日2回投与し、本剤の平均血中トラフ濃度(C0)を3〜8ng/mLに維持するように投与量を調節したときの血中トラフ濃度は以下のとおりであった10)(日本人及び外国人のデータ、分析方法:LC/MS/MS法)。
| 評価時点(移植後) | 本剤の血中トラフ濃度 平均値±標準偏差 |
| 5週 | 4.2±2.20(n=121) |
| 6週 | 4.2±1.94(n=132) |
| 2ヵ月 | 4.6±1.86(n=132) |
| 3ヵ月 | 4.9±2.10(n=134) |
| 4ヵ月 | 5.1±2.46(n=131) |
| 6ヵ月 | 5.0±2.13(n=124) |
| 9ヵ月 | 5.0±2.18(n=121) |
| 12ヵ月 | 5.1±2.12(n=118) |
(2)新規生体肝移植患者にタクロリムスとともに本剤2mg/日を開始用量として1日2回投与した時の移植6ヵ月後の定常状態薬物動態パラメータは以下のとおりで、Cmaxは約15ng/mL、AUCτは約100ng・hr/mL、またPTF(ピーク−トラフ濃度変動)は約110%であった。
| 本剤の薬物動態パラメータ | 本剤1.0mg/回、1日2回投与 |
| 6ヵ月目 | |
| 例数 | 9 |
| Cminss(ng/mL) | 6.30±1.98 |
| Tmax(hr) | 1(1〜8) |
| Cmaxss(ng/mL) | 15.2±4.11 |
| AUCτss(ng・hr/mL) | 101±18.9 |
| Cavgss(ng/mL) | 8.42±1.58 |
| PTF(%) | 111±47.2 |
単回及び反復経口投与時の血中濃度はELISA法、LC/MS法あるいはLC/MS/MS法にて測定した。なお、3〜32ng/mLの濃度範囲では両測定法で測定した濃度はほぼ同等であった10)(日本人8例及び外国人1例のデータ、分析方法:LC/MS/MS法)。
16.2 吸収
腎移植患者に本剤を0.25〜25mgで経口投与したとき、本剤の血中濃度は投与後1〜2時間でピークに達した。また、本剤の血中濃度は、0.25〜15mgの用量範囲では用量に比例して増加した11)(外国人のデータ)。
16.2.1 食事の影響
16.3 分布
16.4 代謝
16.5 排泄
シクロスポリンを投与している腎移植患者に放射標識本剤を単回投与したところ、放射能のほとんど(80%)は糞便中に排泄され、尿中にはごく一部(5%)が排泄された。なお、尿中及び糞便中に未変化体は検出されなかった15)(外国人のデータ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
腎移植患者における移植後の腎機能障害(Clcreaの範囲;11〜107mL/min)は、本剤の薬物動態に影響を及ぼさなかった16)(外国人のデータ)。
16.6.2 肝機能障害患者
中等度の肝機能障害(Child-Pugh分類クラスB)を有する患者8例における本剤の平均AUCは、健康成人8例の平均AUCよりも2倍高かった。AUCは、血清ビリルビン濃度及びプロトロンビン時間と正の相関を示し、血清アルブミン濃度と負の相関を示した。ビリルビン>2mg/dL、プロトロンビン時間>1.3INR(4秒を超える延長)又はアルブミン<3.5g/dLに該当する場合には、本剤のAUCが健康成人よりも高くなる傾向が認められた17)(外国人のデータ)。重度の肝機能障害(Child-Pugh分類クラスC)の影響は検討していないが、本剤のAUCに対する影響は中等度の肝機能障害と同等かそれ以上であると考えられる。[7.5、9.3参照]
16.6.3 小児等
腎移植患者において、患者の年齢(1〜16歳)、体表面積(0.49〜1.92m2)及び体重(11〜77kg)に比例して、本剤のCL/Fが直線的に増加した。定常状態のCL/Fは10.2±3.0L/hr/m2であり、消失半減期は30±11時間であった8)(外国人のデータ)。
16.6.4 高齢者
16〜70歳の腎移植患者において、年齢増加に伴う本剤の経口クリアランスの低下は、1歳あたり0.33%と小さかった18)(外国人のデータ)。
16.7 薬物相互作用
16.7.1 シクロスポリン
健康成人12例を対象として、本剤2mgとシクロスポリンのマイクロエマルジョン製剤(CsA)175mgを単回併用投与したところ、本剤の単独投与時に比べて本剤のAUCは168%(範囲46%〜365%)、Cmaxは82%(範囲25%〜158%)増加した19)(外国人のデータ)。
| 本剤の薬物動態パラメータ | 本剤単独 | CsA併用 |
| Tmax(hr) | 1.0(0.5〜1.0) | 1.0(0.6〜2.5) |
| Cmax(ng/mL) | 11.6±3.3 | 20.5±3.5 |
| AUC(ng・hr/mL) | 74±26 | 193±47 |
| T1/2(hr) | 25.2±8.2 | 29.0±4.6 |
シクロスポリンのマイクロエマルジョン製剤の安定した用量の投与を受けている維持期腎移植患者24例を対象に、プラセボ、本剤0.75mg、2.5mg又は7.5mgを1日1回28日間併用投与したとき、シクロスポリンの薬物動態に対する本剤併用の大きな影響はみられなかった20)(外国人のデータ)。
| シクロスポリンの薬物動態パラメータ | 投与1日目 | 投与28日目 | 比 | 投与1日目 | 投与28日目 | 比 |
| CsA単独 | プラセボ併用 | CsA単独 | 本剤0.75mg併用 | |||
| Tmaxss(hr) | 1.3(1.0〜1.5) | 1.5(1.0〜1.6) | − | 1.5(1.0〜3.7) | 1.5(1.0〜1.6) | − |
| Cminss(ng/mL) | 119±48 | 119±27 | 1.08 | 126±55 | 118±41 | 1.02 |
| Cmaxss(ng/mL) | 1,162±339 | 1,293±317 | 1.17 | 949±201 | 1,210±186 | 1.31 |
| Cavgss(ng/mL) | 326±88 | 375±63 | 1.20 | 310±58 | 368±72 | 1.19 |
| AUCss(ng・hr/mL) | 3,908±1,060 | 4,496±752 | 1.20 | 3,716±691 | 4,419±861 | 1.19 |
| PTF(%) | 324±54 | 310±43 | 0.99 | 280±112 | 301±39 | 1.21 |
| シクロスポリンの薬物動態パラメータ | 投与1日目 | 投与28日目 | 比 | 投与1日目 | 投与28日目 | 比 |
| CsA単独 | 本剤2.5mg併用 | CsA単独 | 本剤7.5mg併用 | |||
| Tmaxss(hr) | 1.5(1.1〜1.5) | 1.5(1.0〜3.1) | − | 1.5(1.0〜1.5) | 1.5(1.0〜2.0) | − |
| Cminss(ng/mL) | 141±40 | 187±21 | 1.40 | 145±30 | 167±68 | 1.12 |
| Cmaxss(ng/mL) | 1,227±180 | 1,705±260 | 1.41 | 1,274±475 | 1,528±309 | 1.28 |
| Cavgss(ng/mL) | 399±60 | 496±55 | 1.26 | 393±89 | 453±118 | 1.17 |
| AUCss(ng・hr/mL) | 4,783±723 | 5,946±660 | 1.26 | 4,715±1,063 | 5,437±1,420 | 1.17 |
| PTF(%) | 275±41 | 305±26 | 1.12 | 280±85 | 312±90 | 1.16 |
16.7.2 HMG-CoA還元酵素阻害剤(高脂血症用剤)
健康成人を対象に本剤2mgとアトルバスタチン20mg又は本剤2mgとプラバスタチン20mgを単回併用投与したとき(各12例)、本剤及びこれらの薬剤の薬物動態に臨床的に重要な影響は認められなかった21)(外国人のデータ)。
16.7.3 タクロリムス
維持期腎移植患者8例を対象とし、本剤3mg/日と標準量のタクロリムス(投与初日〜10日目)あるいは減量したタクロリムス(投与11日目〜3ヵ月;11日目より半量投与)を併用投与したとき、本剤の併用前と併用後でタクロリムスの薬物動態に変化はなかった。また、減量したタクロリムスと併用したときの本剤の薬物動態は、標準量のタクロリムスと併用したときとほぼ同様であった22)(外国人のデータ)。これらの結果より、本剤はタクロリムスの薬物動態にほとんど影響せず、またタクロリムスの減量は本剤の薬物動態に大きく影響しないと考えられた。
16.8 その他
16.8.1 曝露量と急性拒絶反応、有害事象発現率との関係
<心移植>
新規心移植患者を対象に本剤を1.5mg/日(209例)あるいは3mg/日(211例)で1日2回投与したときの移植後6ヵ月間の本剤の平均血中トラフ濃度(C0)は、生検で確認された急性拒絶反応及び血小板減少の発現率に関連していた7)(外国人のデータ)。
| 本剤の平均血中トラフ濃度(C0)(ng/mL) | ≦3.5 | 3.6〜5.3 | 5.4〜7.3 | 7.4〜10.2 | 10.3〜21.8 |
| 急性拒絶反応抑制率 | 65% | 69% | 80% | 85% | 85% |
| 血小板減少(<75,000/mm3) | 5% | 5% | 6% | 8% | 9% |
<腎移植>
(1)曝露量と急性拒絶反応及び有害事象発現率との関係
新規腎移植患者(60例)を対象に、本剤1.5mg/日を開始用量として1日2回投与し、本剤の平均血中トラフ濃度(C0)を3〜8ng/mLに維持するように投与量を調節した。移植後12ヵ月間本剤を投与したときの投与量(中央値)は1.48〜1.50mg/日の範囲で推移し、本剤の平均血中トラフ濃度(C0)は、ほとんど3〜8ng/mLにコントロールされた。平均血中トラフ濃度と治療を要した生検で確認された急性拒絶反応、尿蛋白/クレアチニン比、高コレステロール血症、創傷治癒不良及び移植後糖尿病の発現率に明確な関連性は認められていない9)(日本人のデータ)。
(2)曝露量と急性拒絶反応及び有害事象発現率との関係
新規腎移植患者を対象に本剤を開始用量1.5mg/日(目標血中トラフ濃度3〜8ng/mL、277例)あるいは3mg/日(目標血中トラフ濃度6〜12ng/mL、279例)で1日2回投与したときの移植後12ヵ月間の本剤の平均血中トラフ濃度(C0)は、治療を要した生検で確認された急性拒絶反応、尿蛋白/クレアチニン比、高コレステロール血症、創傷治癒不良及び移植後糖尿病の発現率に関連していた23)(外国人のデータ)。
| 本剤1.5mg/日群及び3mg/日群の併合解析結果 | |||||
| 本剤の平均血中トラフ濃度(C0)(ng/mL) | <3 | 3〜6 | 6〜8 | 8〜12 | ≧12 |
| 治療を要した急性拒絶反応抑制率(n=547) | 64% | 86% | 85% | 86% | 91% |
| 尿蛋白/クレアチニン比(≧300mg/g)(n=480) | 73% | 48% | 56% | 59% | 92% |
| 高コレステロール血症(≧6.2mmol/L)(n=544) | 77% | 64% | 64% | 77% | 84% |
| 創傷治癒不良(n=529) | 57% | 28% | 28% | 38% | 64% |
| 移植後糖尿病(n=543) | 0% | 7% | 11% | 13% | 20% |
<肝移植>
(1)曝露量と急性拒絶反応及び有害事象発現率との関係
生体肝移植患者(142例)を対象に、本剤2mg/日を開始用量として1日2回投与し、本剤の平均血中トラフ濃度(C0)を3〜8ng/mLに維持するように投与量を調節した。移植後12ヵ月間本剤を投与したときの投与量(中央値)は2.00〜2.50mg/日の範囲で推移し、本剤の平均血中トラフ濃度(C0)は、ほとんど3〜8ng/mLにコントロールされた。平均血中トラフ濃度と治療を要した生検で確認された急性拒絶反応、尿蛋白/クレアチニン比、高コレステロール血症、創傷治癒不良及び移植後糖尿病の発現率に明確な関連性は認められていない10)(日本人及び外国人のデータ)。
(2)曝露量と急性拒絶反応及び有害事象発現率との関係
脳死肝移植患者(245例)を対象に本剤を2mg/日で1日2回投与開始し、本剤の平均血中トラフ濃度(C0)を3〜8ng/mLに維持するように投与量を調節したときの移植後12ヵ月間の本剤の平均血中トラフ濃度(C0)は、ほとんどは3〜8ng/mLにコントロールされており、治療を要した生検で確認された急性拒絶反応、尿蛋白/クレアチニン比、高コレステロール血症、創傷治癒不良及び移植後糖尿病の発現率に関連性は認められていない2)(外国人のデータ)。
17. 臨床成績
17.1 有効性及び安全性に関する試験
<心移植>
17.1.1 海外第III相試験(B253試験)
新規心移植患者を対象とした本試験では、標準量シクロスポリンのマイクロエマルジョン製剤(CsA)及び副腎皮質ホルモン剤と併用した本剤(固定用量1.5mg/日及び3mg/日)の有効性及び安全性を、標準量CsAと併用したアザチオプリン(AZA)1〜3mg/kg/日と比較した。ISHLT基準によるグレード3A以上の急性拒絶反応、血行動態異常を伴う急性拒絶反応、移植心廃絶、死亡又は追跡調査不能からなる複合評価項目の6ヵ月後の発現率を主要評価項目とした。
| 本剤(固定用量1.5mg/日)+標準量CsA群 (n=209) | 本剤(固定用量3.0mg/日)+標準量CsA群 (n=211) | AZA+標準量CsA群 (n=214) | p値 | |
| 効果不十分(6ヵ月) | 76(36.4%) | 57(27.0%) | 100(46.7%) | 0.031a <0.001b 0.037c |
| 生検で確認されたグレード3A(ISHLT)以上の急性拒絶反応 | 58(27.8%) | 40(19.0%) | 89(41.6%) | 0.003a <0.001b 0.032c |
| 血行動態異常を伴う急性拒絶反応の臨床的疑い | 14(6.7%) | 11(5.2%) | 16(7.5%) | n.s. |
| 移植心廃絶 | 4(1.9%) | 8(3.8%) | 6(2.8%) | n.s. |
| 死亡 | 13(6.2%) | 14(6.6%) | 12(5.6%) | n.s. |
| 追跡調査不能 | 0 | 0 | 1(0.5%) | n.s. |
なお、シクロスポリンの用量は、血中トラフ濃度(C0)が以下の目標範囲内に収まるように調節した:1〜4週:250〜400ng/mL、1〜6ヵ月:200〜350ng/mL、7〜24ヵ月:100〜300ng/mL。
実際の血中トラフ濃度(C0)を以下の表に示す。
実際の血中トラフ濃度(C0)を以下の表に示す。
| 本剤の投与期間 | シクロスポリンの血中トラフ濃度(C0)(ng/mL) | |
| 本剤(固定用量1.5mg/日)+標準量CsA群 | 本剤(固定用量3mg/日)+標準量CsA群 | |
| 1週目 | 220±120 n=167 | 226±132 n=167 |
| 1ヵ月目 | 270±119 n=163 | 255±111 n=159 |
| 6ヵ月目 | 201±109 n=112 | 185±87 n=115 |
| 12ヵ月目 | 166±84 n=99 | 148±70 n=94 |
| 24ヵ月目 | 150±92 n=65 | 137±55 n=58 |
また、血清クレアチニン値上昇の発現率は、AZA+標準量CsA群の患者よりも本剤を標準量CsAと併用した患者の方が高値を示した。この所見から、本剤はシクロスポリンによって誘発される腎毒性を増強することが示唆される。この影響は、シクロスポリンを減量することによって回復すると考えられるが、シクロスポリンの血中トラフ濃度(C0)が最初の3ヵ月間に175ng/mLを下回る場合、6ヵ月後に135ng/mLを下回る場合及び6ヵ月後以降に100ng/mLを下回る場合の心移植患者への本剤の投与については限られたデータしかない24)。
副作用発現頻度は、本剤1.5mg群で69.9%(146/209例)及び本剤3mg群で73.0%(154/211例)であった(24ヵ月の集計)。主な副作用は、本剤1.5mg群では白血球減少症42例(20.1%)、高脂血症18例(8.6%)、血小板減少症16例(7.7%)、腎機能障害16例(7.7%)、本剤3mg群で白血球減少症41例(19.4%)、血小板減少症29例(13.7%)、高脂血症24例(11.4%)であった。
<腎移植>
17.1.2 国内第III相試験(A1202試験)
新規腎移植患者を対象とした本試験では、減量シクロスポリンのマイクロエマルジョン製剤(CsA)と併用した本剤(開始用量1.5mg/日、(目標血中トラフ濃度3〜8ng/mLを維持するように投与量を調整)の有効性及び安全性を、標準量CsAと併用したミコフェノール酸モフェチル(MMF)2g/日と比較した。なおいずれも副腎皮質ホルモン剤及びバシリキシマブを施設方法に準じて併用した。治療を要し生検で確認された急性拒絶反応(Banff97分類による)、移植腎廃絶、死亡又は追跡調査不能からなる複合評価項目(効果不十分)の移植12ヵ月後の発現率を主要評価項目とした。
| 本剤(開始用量1.5mg/日)+減量CsA群 (n=61) | MMF+標準量CsA群 (n=61) | |
| n(%) | ||
| 効果不十分(12ヵ月) | 7(11.5) | 7(11.5) |
| 治療を要し生検で確認された急性拒絶反応 | 3(4.9) | 5(8.2) |
| 移植腎廃絶 | 0(0.0) | 0(0.0) |
| 死亡 | 0(0.0) | 0(0.0) |
| 追跡調査不能 | 4(6.6) | 2(3.3) |
なお、CsAの用量は、シクロスポリンの血中トラフ濃度(C0)が以下の目標範囲に収まるように調節した。移植後5日〜1ヵ月:100〜200ng/mL、2〜3ヵ月:75〜150ng/mL、4〜5ヵ月:50〜100ng/mL、6ヵ月以降:25〜50ng/mL。
実際の血中トラフ濃度(C0)を以下の表に示す9)。
実際の血中トラフ濃度(C0)を以下の表に示す9)。
| 本剤の投与期間 | シクロスポリンの血中トラフ濃度(C0)(ng/mL)、本剤(開始用量1.5mg/日)+減量CsA群 |
| 1週目 | 247.5(107〜512)n=59 |
| 1ヵ月目 | 191.0(90〜450)n=55 |
| 3ヵ月目 | 130.0(57〜301)n=54 |
| 6ヵ月目 | 104.5(43〜221)n=54 |
| 9ヵ月目 | 73.0(0〜146)n=53 |
| 12ヵ月目 | 63.0(0〜145)n=52 |
副作用発現頻度は、本剤群で95.1%(58/61例)であった(12ヵ月の集計)。主な副作用は、高脂血症26例(42.6%)、鼻咽頭炎18例(29.5%)、発熱及び高血圧各13例(21.3%)であった。
17.1.3 海外第III相試験(A2309試験)
新規腎移植患者を対象とした本試験では、減量シクロスポリンのマイクロエマルジョン製剤(CsA)と併用した本剤(開始用量1.5mg/日(目標血中トラフ濃度3〜8ng/mLを維持するように投与量を調整)及び開始用量3mg/日(目標血中トラフ濃度6〜12ng/mLを維持するように投与量を調整))の有効性及び安全性を、標準量CsAと併用したミコフェノール酸ナトリウム腸溶錠(Myfortic:国内未承認)1.44g/日と比較した。なおいずれも副腎皮質ホルモン剤及びバシリキシマブを施設方法に準じて併用した。主要な評価項目は国内試験(A1202試験)と同じである。
| 本剤(開始用量1.5mg/日)+減量CsA群 (n=277) | 本剤(開始用量3mg/日)+減量CsA群 (n=279) | Myfortic+標準量CsA群 (n=277) | |
| n(%) | |||
| 効果不十分(12ヵ月) | 75(27.1) | 60(21.5) | 70(25.3) |
| 治療を要し生検で確認された急性拒絶反応 | 48(17.3) | 38(13.6) | 50(18.1) |
| 移植腎廃絶 | 13(4.7) | 13(4.7) | 9(3.2) |
| 死亡 | 8(2.9) | 9(3.2) | 6(2.2) |
| 追跡調査不能 | 12(4.3) | 6(2.2) | 9(3.2) |
シクロスポリンの血中トラフ濃度(C0)の目標範囲は国内試験(A1202試験)と同じである。
実際の血中トラフ濃度(C0)を以下の表に示す23)。
実際の血中トラフ濃度(C0)を以下の表に示す23)。
| 本剤の投与期間 | シクロスポリンの血中トラフ濃度(C0)(ng/mL) | |
| 本剤(開始用量1.5mg/日)+減量CsA群 | 本剤(開始用量3mg/日)+減量CsA群 | |
| 1週目 | 185.0(13〜571)n=265 | 179.0(20〜596)n=266 |
| 1ヵ月目 | 161.5(32〜705)n=244 | 162.0(25〜887)n=245 |
| 3ヵ月目 | 110.0(25〜469)n=221 | 112.0(25〜808)n=218 |
| 6ヵ月目 | 75.0(12〜502)n=200 | 74.5(24〜241)n=188 |
| 9ヵ月目 | 48.3(20〜136)n=196 | 43.0(19〜274)n=192 |
| 12ヵ月目 | 45.5(11〜350)n=191 | 43.5(20〜176)n=180 |
副作用発現頻度は、本剤1.5mg群で70.4%(193/274例)及び本剤3mg群で76.3%(212/278例)であった(24ヵ月の集計)。主な副作用は、本剤1.5mg群では高コレステロール血症14.6%(40/274例)、脂質異常症14.2%(39/274例)、高脂血症13.9%(38/274例)、本剤3mg群で高脂血症16.9%(47/278例)、高コレステロール血症15.5%(43/278例)、脂質異常症11.9%(33/278例)であった。
<肝移植>
17.1.4 国際共同第III相試験(H2307試験)
生体肝移植患者を対象とした本試験では、減量タクロリムスと併用した本剤(開始用量2.0mg/日、目標血中トラフ濃度3〜8ng/mLを維持するように投与量を調整)の有効性及び安全性を、標準量タクロリムスと比較した。なお本剤は移植後4週から投与を開始した。いずれも副腎皮質ホルモン剤及びMMF、バシリキシマブを施設方法に準じて併用し、MMFの投与は移植後4週までとした。治療を要し生検で確認された急性拒絶反応(Banff97分類による)、移植肝廃絶、死亡からなる複合評価項目(効果不十分)の移植12ヵ月後の発現率を主要評価項目とした。
| 本剤(開始用量2.0mg/日)+減量タクロリムス群 (n=142) | 標準量タクロリムス群 (n=142) | |
| n(%) | ||
| 効果不十分(12ヵ月) | 7(4.9) | 8(5.6) |
| 治療を要し生検で確認された急性拒絶反応 | 3(2.1) | 5(3.5) |
| 移植肝廃絶 | 0(0) | 0(0) |
| 死亡 | 4(2.8) | 3(2.1) |
なお、タクロリムスの用量は、タクロリムスの血中トラフ濃度(C0)が以下の目標範囲に収まるように調節した。移植後1ヵ月以降:3〜5ng/mL。
実際の血中トラフ濃度(C0)を以下の表に示す10)。
実際の血中トラフ濃度(C0)を以下の表に示す10)。
| 移植後の経過時点 | タクロリムスの血中トラフ濃度(C0)(ng/mL)、本剤(開始用量2.0mg/日)+減量タクロリムス群 |
| 5週目(本剤の投与から1週目) | 6.3(1.0〜21.3)n=125 |
| 2ヵ月目 | 4.8(1.9〜13.0)n=131 |
| 3ヵ月目 | 5.2(1.3〜11.3)n=133 |
| 6ヵ月目 | 4.6(0.7〜12.5)n=126 |
| 9ヵ月目 | 4.0(1.0〜10.2)n=122 |
| 12ヵ月目 | 4.3(0.1〜10.1)n=121 |
副作用発現頻度は、日本人15例を含む本剤群で57.0%(81/142例)であった(12ヵ月の集計)。主な副作用は、白血球減少症12例(8.5%)、高脂血症12例(8.5%)、高コレステロール血症10例(7.0%)であった。
17.1.5 海外第III相試験(H2304試験)
脳死肝移植患者を対象とした本試験では、減量タクロリムスと併用した本剤(開始用量2.0mg/日、目標血中トラフ濃度3〜8ng/mLを維持するように投与量を調整)の有効性及び安全性を、標準量タクロリムスと比較した。なお本剤は移植後4週から投与を開始した。いずれも副腎皮質ホルモン剤及びMMFを医療機関での方法に準じて併用し、MMFの投与は移植後4週までとした。主要な評価項目は生体肝移植臨床試験(H2307試験)と同じである。
| 本剤(開始用量2.0mg/日)+減量タクロリムス群 (n=245) | 標準量タクロリムス群 (n=243) | |||
| 12ヵ月 | 24ヵ月 | 12ヵ月 | 24ヵ月 | |
| n(%) | ||||
| 効果不十分 | 16(6.5) | 24(9.8) | 23(9.5) | 29(11.9) |
| 治療を要し生検で確認された急性拒絶反応 | 7(2.9) | 11(4.5) | 17(7.0) | 18(7.4) |
| 移植肝廃絶 | 6(2.4) | 9(3.7) | 3(1.2) | 7(2.9) |
| 死亡 | 9(3.7) | 12(4.9) | 6(2.5) | 10(4.1) |
なお、本剤(開始用量2.0mg/日)+減量タクロリムス群でのタクロリムスの血中トラフ濃度(C0)の目標範囲は生体肝移植臨床試験(H2307試験)と同じである。
実際の血中トラフ濃度(C0)を以下の表に示す2)。
実際の血中トラフ濃度(C0)を以下の表に示す2)。
| 移植後の経過時点 | タクロリムスの血中トラフ濃度(C0)(ng/mL)、本剤(開始用量2.0mg/日)+減量タクロリムス群 |
| 5週目(本剤の投与から1週目) | 8.2(1.7〜26.5)n=184 |
| 2ヵ月目 | 5.7(2.0〜28.0)n=186 |
| 3ヵ月目 | 5.2(1.2〜18.7)n=186 |
| 6ヵ月目 | 4.8(2.0〜15.1)n=160 |
| 9ヵ月目 | 4.8(1.9〜16.1)n=151 |
| 12ヵ月目 | 4.5(0.8〜10.6)n=135 |
| 18ヵ月目 | 4.4(1.0〜10.8)n=127 |
| 24ヵ月目 | 3.8(0.7〜15.3)n=109 |
副作用発現頻度は、本剤群で70.6%(173/245例)であった(24ヵ月の集計)。主な副作用は、白血球減少症22例(9.0%)、高血圧21例(8.6%)、高脂血症18例(7.3%)、腎不全18例(7.3%)であった。
18. 薬効薬理
18.1 作用機序
18.1.1 FKBP12に結合した25)。
18.2 In vitro免疫抑制作用
18.2.1 マウス及びヒトの混合リンパ球反応(MLR)を阻害し、同種抗原により誘導されるT細胞の増殖を抑制した28)。
18.2.2 Trinitrophenyl-lipopolysaccharide、N-2,4-dinitrophenyl-β-Ala-Gly-Gly-AECM-Ficoll及びヒツジ赤血球(SRBC)に対するマウスB細胞の免疫反応を抑制した29)。
18.3 In vivo免疫抑制作用
18.3.1 ラット後足蹠への同種ドナー脾細胞の皮下注入により誘発した局所移植片対宿主反応(膝窩リンパ節の腫脹)を抑制した25)。
18.4 慢性拒絶反応抑制作用
18.5 シクロスポリンとの相乗効果
18.5.1 エベロリムスとシクロスポリンの併用により、マウスのMLRが相乗的に阻害された34)。
18.6 再発狭窄抑制作用
19. 有効成分に関する理化学的知見
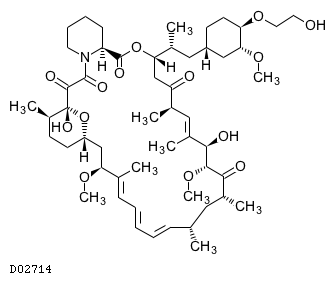
19.1. エベロリムス
| 一般的名称 | エベロリムス |
|---|---|
| 一般的名称(欧名) | Everolimus |
| 化学名 | (1R,9S,12S,15R,16E,18R,19R,21R,23S,24E,26E,28E,30S,32S,35R)-1,18-Dihydroxy-12-{(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxycyclohexyl]-1-methylethyl}-19,30-dimethoxy-15,17,21,23,29,35-hexamethyl-11,36-dioxa-4-azatricyclo[30.3.1.04,9]hexatriaconta-16,24,26,28-tetraene-2,3,10,14,20-pentaone |
| 分子式 | C53H83NO14 |
| 分子量 | 958.22 |
| 物理化学的性状 | 白色〜淡黄色の粉末で、エタノール(99.5)に溶けやすく、水にほとんど溶けない。 |
| KEGG DRUG |
|
20. 取扱い上の注意
光及び湿気を避けるため、PTP包装のまま保存すること。
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
<心移植における拒絶反応の抑制>
国内での治験は実施されておらず、患者数が極めて限られていることから、製造販売後、一定数の症例に係るデータが集積されるまでの間は、全症例を対象に使用成績調査を実施することにより、本剤使用患者の背景情報を把握するとともに、本剤の安全性及び有効性に関するデータを早期に収集し、本剤の適正使用に必要な措置を講じること。
22. 包装
<サーティカン錠0.25mg>
60錠[10錠(両面アルミニウムPTP)×6]
<サーティカン錠0.5mg>
60錠[10錠(両面アルミニウムPTP)×6]
<サーティカン錠0.75mg>
60錠[10錠(両面アルミニウムPTP)×6]
23. 主要文献
- Johnson,R.W.G.et al., Transplantation., 72 (5), 777-786, (2001) »PubMed »DOI
- 社内資料:外国人脳死肝移植患者を対象とした比較対照試験(H2304試験)(2018年2月23日承認、CTD2.7.6-4.1.2)
- 社内資料:心移植患者及び腎移植患者を対象とした製造販売後調査(A1401調査及びA1402調査)
- 社内資料:小児肝移植患者を対象とした海外臨床試験(H2305試験)(2018年2月23日承認、CTD2.7.6-4.2.1)
- 社内資料:健康成人(日本人)に対する臨床第I相試験(単回投与)(2007年1月26日承認、CTD2.7.6-4 試験番号1101)
- 社内資料:新規腎移植患者における3剤併用免疫抑制療法の有効性及び安全性についての検討(2007年1月26日承認、CTD2.7.6-5 試験番号B251)
- Kovarik,J.M.et al., J.Heart Lung Transplant., 22 (10), 1117-1125, (2003) »PubMed »DOI
- 社内資料:小児腎移植患者における薬物動態、安全性及び忍容性の検討(2007年1月26日承認、CTD2.7.6-5 試験番号B257,試験番号B351)
- 社内資料:日本人新規腎移植患者を対象とした比較対照試験(2011年12月22日承認、CTD2.7.6-4.1.1)(A1202試験)
- 社内資料:生体肝移植患者を対象とした国際共同試験(H2307試験)(2018年2月23日承認、CTD2.7.6-4.1.1)
- 社内資料:維持期腎移植患者における安全性及び忍容性の検討(2007年1月26日承認、CTD2.7.6-8 試験番号W101)
- Kovarik,J.M.et al., Pharmacotherapy., 22 (2), 154-159, (2002) »PubMed »DOI
- 社内資料:[3H]-エベロリムスの血中分布に関する検討(2007年1月26日承認、CTD2.7.2-3.5)
- 社内資料:In vitro代謝(2007年1月26日承認、CTD2.6.4-5.2)
- 社内資料:維持期腎移植患者における[14C]-エベロリムス単回経口投与後の吸収、分布、動態及び生体内変換についての検討(2007年1月26日承認、CTD2.7.6-5 試験番号W107)
- 社内資料:新規腎移植患者における安全性、忍容性及び薬物動態の検討(1年の検討)(2007年1月26日承認、CTD2.7.6-14 試験番号B157)
- Kovarik,J.M.et al., Clin.Pharmacol.Ther., 70 (5), 425-430, (2001) »PubMed
- 社内資料:新規腎移植患者における母集団薬物動態解析(2007年1月26日承認、CTD2.7.2-4.3)
- Kovarik,J.M.et al., J.Clin.Pharmacol., 42 (1), 95-99, (2002) »PubMed »DOI
- 社内資料:維持期腎移植患者における安全性、忍容性及び薬物動態の検討(2007年1月26日承認、CTD2.7.6-8 試験番号B154)
- Kovarik,J.M.et al., J.Clin.Pharmacol., 42 (2), 222-228, (2002) »PubMed »DOI
- Kovarik,J.M.et al., Transplant.Proc., 38 (10), 3456-3458, (2006) »PubMed »DOI
- 社内資料:外国人新規腎移植患者を対象とした比較対照試験(A2309試験)(2011年12月22日承認、CTD2.7.6-4.1.2)
- 社内資料:新規心移植患者における有効性及び安全性の検討(B253試験)(2007年1月26日承認、CTD2.7.6-9.1 試験番号B253)
- Schuler,W.et al., Transplantation., 64 (1), 36-42, (1997) »PubMed »DOI
- 社内資料:増殖因子によって刺激されたp70S6キナーゼ活性に対する阻害作用(2007年1月26日承認、CTD2.6.2-2.1.4)
- 社内資料:ラット末梢血リンパ球におけるp70S6キナーゼ活性(単回投与)(2007年1月26日承認、CTD2.6.2-2.1.4)
- 社内資料:マウス及びヒトT細胞の増殖に対する作用(2007年1月26日承認、CTD2.6.2-2.1.1,2.2.1)
- 社内資料:T細胞非依存性抗原及びT細胞依存性抗原に対するマウスのB細胞応答に及ぼす作用(2007年1月26日承認、CTD2.6.2-2.2.3,2.3.2)
- 社内資料:カニクイザルにおけるA及びB型肝炎ワクチン接種に対する作用(2007年1月26日承認、CTD2.6.2-2.3.2)
- Schuurman,H.J.et al., Transplantation., 69 (5), 737-742, (2000) »PubMed »DOI
- 社内資料:ラットの大動脈移植モデルに対する作用(2007年1月26日承認、CTD2.6.2-2.4.5)
- Schuurman,H.J.et al., Transplant.Proc., 31 (1-2), 1024-1025, (1999) »PubMed »DOI
- Schuurman,H.J.et al., Transplantation., 64 (1), 32-35, (1997) »PubMed »DOI
- Hausen,B.et al., Transplantation., 67 (7), 956-962, (1999) »PubMed »DOI
- Hausen,B.et al., Transplantation., 69 (1), 76-86, (2000) »PubMed »DOI
- 社内資料:ラット頚動脈バルーン傷害モデル(14日間)における内膜肥厚抑制作用(2007年1月26日承認、CTD2.6.2-3.2)
- 社内資料:ラット頚動脈バルーン傷害モデル(28日間)における内膜肥厚抑制作用(2007年1月26日承認、CTD2.6.2-3.2)
- 社内資料:ブタ冠動脈傷害(PTCA)モデル(14日間)における内膜肥厚抑制作用(2007年1月26日承認、CTD2.6.2-3.2)
- Farb,A.et al., Circulation., 106 (18), 2379-2384, (2002) »PubMed »DOI
24. 文献請求先及び問い合わせ先
文献請求先
ノバルティスファーマ株式会社
ノバルティスダイレクト
〒105-6333
東京都港区虎ノ門1-23-1
電話:0120-003-293 受付時間:月〜金 9:00〜17:30(祝祭日及び当社休日を除く)
製品情報問い合わせ先
ノバルティスファーマ株式会社
ノバルティスダイレクト
〒105-6333
東京都港区虎ノ門1-23-1
電話:0120-003-293 受付時間:月〜金 9:00〜17:30(祝祭日及び当社休日を除く)
26. 製造販売業者等
26.1 製造販売
ノバルティスファーマ株式会社
東京都港区虎ノ門1-23-1