医薬品情報
| 総称名 | チャンピックス |
|---|---|
| 一般名 | バレニクリン酒石酸塩 |
| 欧文一般名 | Varenicline Tartrate |
| 製剤名 | バレニクリン酒石酸塩錠 |
| 薬効分類名 | α4β2ニコチン受容体部分作動薬(禁煙補助薬) |
| 薬効分類番号 | 7990 |
| ATCコード | N07BA03 |
| KEGG DRUG |
D06282
バレニクリン酒石酸塩
|
| JAPIC | 添付文書(PDF) |
添付文書情報2020年10月 改訂(第1版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| チャンピックス錠0.5mg | CHAMPIX Tablets 0.5mg | ファイザー | 7990003F1028 | 77.5円/錠 | 劇薬, 処方箋医薬品 |
| チャンピックス錠1mg | CHAMPIX Tablets 1mg | ファイザー | 7990003F2024 | 136.1円/錠 | 劇薬, 処方箋医薬品 |
2. 禁忌
次の患者には投与しないこと
本剤の成分に対し過敏症の既往歴のある患者
4. 効能または効果
ニコチン依存症の喫煙者に対する禁煙の補助
5. 効能または効果に関連する注意
5.1 ニコチン依存症の診断については、ニコチン依存症に係わるスクリーニングテスト(TDS)により診断すること1)。
5.2 本剤の使用にあたっては、患者に禁煙意志があることを確認すること。
6. 用法及び用量
通常、成人にはバレニクリンとして第1〜3日目は0.5mgを1日1回食後に経口投与、第4〜7日目は0.5mgを1日2回朝夕食後に経口投与、第8日目以降は1mgを1日2回朝夕食後に経口投与する。なお、本剤の投与期間は12週間とする。
7. 用法及び用量に関連する注意
7.1 本剤は原則として、他の禁煙補助薬と併用しないこと。本剤の有効性及び安全性は単剤投与により確認されており、他の禁煙補助薬と併用した際の有効性は検討されておらず、安全性についても経皮吸収ニコチン製剤との併用時に副作用発現率の上昇が認められている。[16.7.3参照]
7.2 患者が禁煙を開始する日を設定すること。その日から1週間前に本剤の投与を始めること。
7.3 本剤による12週間の禁煙治療により禁煙に成功した患者に対して、長期間の禁煙をより確実にするために、必要に応じ、本剤をさらに延長して投与することができる。その場合にはバレニクリンとして1mgを1日2回、朝夕食後に12週間投与すること。[17.1.5参照]
7.4 最初の12週間の投与期間中に禁煙に成功しなかった患者や投与終了後に再喫煙した患者で、再度本剤を用いた禁煙治療を実施する場合には、過去の禁煙失敗の要因を明らかにし、それらの要因への対処を行った後のみに、本剤の投与を開始すること。
7.5 本剤の忍容性に問題がある場合には、0.5mg1日2回に減量することができる。
8. 重要な基本的注意
8.1 医師等により、禁煙治療プログラムに基づいた指導の下に本剤を適切に使用すること。
8.2 禁煙は治療の有無を問わず様々な症状(不快、抑うつ気分、不眠、いらだたしさ、欲求不満、怒り、不安、集中困難、落ち着きのなさ、心拍数の減少、食欲増加、体重増加等)を伴うことが報告されており2)、基礎疾患として有している精神疾患の悪化を伴うことがある。
8.3 抑うつ気分、不安、焦燥、興奮、行動又は思考の変化、精神障害、気分変動、攻撃的行動、敵意、自殺念慮及び自殺が報告されている。本剤との因果関係は明らかではないが、これらの症状があらわれることがあるので、本剤を投与する際には患者の状態を十分に観察すること。なお、本剤中止後もこれらの症状があらわれることがある。また、これらの症状・行動があらわれた場合には本剤の服用を中止し、速やかに医師等に連絡するよう患者に指導すること。
8.4 めまい、傾眠、意識障害等があらわれ、自動車事故に至った例も報告されているので、自動車の運転等危険を伴う機械の操作に従事させないよう注意すること。[11.1.3参照]
8.5
本剤の投与の有無にかかわらず、禁煙により生じる生理的な変化のため、下記のような薬剤の薬物動態や薬力学が変化し、用量調節が必要になる場合がある。
テオフィリン、ワルファリン、インスリン等
また、喫煙によりCYP1A2の活性が誘導されるため、禁煙を開始後、CYP1A2の基質となる薬剤の血漿濃度が上昇する可能性がある。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 統合失調症、双極性障害、うつ病等の精神疾患のある患者
精神症状を悪化させることがある。
9.2 腎機能障害患者
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。バレニクリン15mg/kg/日をラットの妊娠〜授乳期間中に経口投与したところ、出生児に体重及び受胎能の低下と聴覚性驚愕反応の亢進が認められた。また、妊娠ウサギにバレニクリン30mg/kg/日を経口投与したところ、胎児の体重低下が認められた。
9.6 授乳婦
授乳中の女性には、治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。ヒト母乳中への本剤の移行は不明であるが、動物実験(ラット)で乳汁中に移行することが報告されている。
9.7 小児等
国内では小児等を対象とした臨床試験は実施していない。
9.8 高齢者
10. 相互作用
10.2 併用注意
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 皮膚粘膜眼症候群(Stevens-Johnson症候群)(頻度不明)、多形紅斑(頻度不明)
皮疹等の症状があらわれた場合には投与を中止し、適切な処置を行うこと。
11.1.2 血管浮腫(頻度不明)
顔面、舌、口唇、咽頭、喉頭等の腫脹を症状とする血管浮腫があらわれることがある。
11.1.3 意識障害(頻度不明)
意識レベルの低下、意識消失等の意識障害があらわれることがある。[8.4参照]
11.1.4 肝機能障害(頻度不明)、黄疸(頻度不明)
AST、ALT、γ-GTP等の上昇を伴う肝機能障害、黄疸があらわれることがある。
発現頻度は、承認時の国内及び外国第II相/第III相試験の結果に基づいている。
11.2 その他の副作用
| 5%以上 | 0.5%以上5%未満 | 0.5%未満 | 頻度不明 | |
| 感染症及び寄生虫症 | 上気道感染 | 気管支炎 | ||
| 代謝及び栄養障害 | 食欲不振、食欲亢進 | 多飲症 | ||
| 精神障害 | 不眠症(16.3%)、異常な夢(13.0%) | リビドー減退、易刺激性、感情不安定、激越、睡眠障害、不安、抑うつ、落ち着きのなさ | 精神緩慢、気分変動、思考異常、不快気分 | 精神障害、攻撃的行動、敵意 |
| 神経系障害 | 頭痛(11.6%) | 傾眠、振戦、注意力障害、味覚異常、嗜眠 | 協調運動異常、構語障害、感覚鈍麻 | 記憶障害、健忘、一過性健忘、痙攣 |
| 心臓障害 | 心房細動、動悸、狭心症 | |||
| 血管障害 | ほてり、高血圧 | |||
| 眼障害 | 眼痛、羞明、暗点、結膜炎 | |||
| 耳及び迷路障害 | 耳鳴 | |||
| 呼吸器、胸郭及び縦隔障害 | 咽喉刺激感、咳嗽 | 呼吸困難、嗄声、鼻漏、気道うっ血、副鼻腔うっ血、いびき | ||
| 胃腸障害 | 嘔気(28.5%)、鼓腸、便秘 | 胃食道逆流性疾患、胃不快感、下痢、口内乾燥、消化不良、軟便、腹痛、腹部膨満、嘔吐 | おくび、胃炎、歯肉痛、吐血、血便排泄、口内炎 | イレウス |
| 皮膚及び皮下組織障害 | ざ瘡、そう痒症、発疹 | 紅斑、多汗症 | ||
| 筋骨格系及び結合組織障害 | 筋痛、筋痙攣 | 関節硬直、関節痛、背部痛 | ||
| 腎及び尿路障害 | 頻尿・夜間頻尿 | 糖尿、多尿 | ||
| 生殖系及び乳房障害 | 月経過多、性機能不全 | |||
| 全身障害及び投与局所様態 | 胸痛、倦怠感、口渇、無力症、めまい | 胸部不快感、発熱 | 浮腫、末梢性浮腫 | |
| 臨床検査 | 肝機能検査値異常(AST上昇、ALT上昇、ALP上昇、血中ビリルビン上昇) | 心電図ST部分下降、心電図T波振幅減少、心拍数増加、血小板数減少、体重増加 |
13. 過量投与
13.1 処置
過量投与後の透析の臨床経験はないが、バレニクリンは透析により除去されることが示されている。[16.6.1参照]
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
15. その他の注意
15.1 臨床使用に基づく情報
15.1.1 海外で実施された心血管疾患を有する患者703例を対象とした本剤の有効性評価のためのランダム化二重盲検比較試験において、全試験期間における心血管イベント(心血管死、冠動脈疾患、脳血管疾患、末梢血管疾患)の発生割合は本剤投与群では7.1%(25/353)、プラセボ投与群では5.7%(20/350)[リスク差:1.4%、95%信頼区間−2.3%〜5.0%]であったとの報告がある3)。また、上記試験を含む15のランダム化二重盲検比較試験の心血管イベント発生に関する安全性メタ解析において、投与期間及び投与期間+30日における主要心血管イベント(心血管死、非致死性心筋梗塞、非致死性脳卒中)の発生割合及びハザード比は以下の通りであった。
投与期間[発生割合:本剤投与群0.17%(7/4190)、プラセボ投与群0.07%(2/2812)、ハザード比:2.83、95%信頼区間0.76〜10.55]4)。
なお、安全性メタ解析に用いた主要心血管イベントは、主として心血管疾患を有する等の高リスク患者で起きたものである。
15.1.2 海外で実施された12〜19歳の喫煙者312例を対象とした本剤の有効性・安全性評価のための無作為化二重盲検プラセボ対照比較試験において、主要評価項目の投与開始後第9〜12週の4週間持続禁煙率は、バレニクリン群がプラセボ群と比較して統計学的に有意な増加を示さなかった。
15.2 非臨床試験に基づく情報
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
健康成人男性喫煙者14例にバレニクリン0.25、0.5、1及び2mgを食後単回投与した時の最高血漿中濃度(Cmax)はそれぞれ1.32、2.45、4.97及び9.96ng/mL、血漿中濃度−時間曲線下面積(AUC)はそれぞれ26.2、50.0、104及び226ng・h/mLであり、用量の増加に伴い増加した。最高血漿中濃度到達時間(Tmax)の平均値はいずれの投与量においても約3時間であった。血漿中濃度半減期(t1/2)の平均値は0.25、0.5、1及び2mg投与に対し、それぞれ13.1、14.5、18.4及び19.3時間であった9)。
| 投与量(mg) | n | Cmax(ng/mL) | Tmax(h) | t1/2(h) | AUC(ng・h/mL) |
| 0.25 | 12 | 1.32±0.11 | 2.75±1.06 | 13.1±2.10 | 26.2±3.88 |
| 0.5 | 11 | 2.45±0.24 | 2.36±0.92 | 14.5±2.40 | 50.0±5.88 |
| 1 | 12 | 4.97±0.56 | 2.75±0.75 | 18.4±3.15 | 104±10.8 |
| 2 | 11 | 9.96±1.25 | 3.09±1.38 | 19.3±2.17 | 226±46.9 |
注)本剤の承認用量は1回1mgまでである。
16.1.2 反復投与
健康成人男性喫煙者12例にバレニクリン0.5及び1mg1日2回を14日間反復経口投与した時、バレニクリン濃度は投与4日目には定常状態に達し、単回投与試験の結果から予想される蓄積を上回る値は認められなかった10)。
| 薬物動態パラメータ | 0.5mg投与群(n=8) | 1mg投与群(n=8) | ||
| 投与1日目 | 投与14日目 | 投与1日目 | 投与14日目 | |
| AUCτa)(ng・h/mL) | 21.79±3.02 | 58.48±10.38 | 42.68±6.14 | 116.00±29.27 |
| Cmaxa)(ng/mL) | 2.62±0.32 | 5.94±1.06 | 5.29±0.89 | 11.95±2.86 |
| Tmaxa)(h) | 3.13±0.99 | 3.50±0.93 | 2.50±0.93 | 3.13±0.64 |
| t1/2(h) | NA | 27.98±4.52 | NA | 24.21±3.46 |
| Racτa) | 2.700±0.400 | 2.697±0.316 | ||
| CLr(mL/min) | 79.02±14.84 | 83.70±14.86 | 99.25±23.66 | 90.46±19.97 |
16.2 吸収
16.2.1 食事の影響
健康成人喫煙者12例にバレニクリン1mgを空腹時及び食後に単回経口投与し、薬物動態を比較した。Cmax及びAUCは空腹時投与と食後投与の間で同等の値を示したことから、バレニクリンの薬物動態に対する食事の影響はない11)(外国人データ)。
16.3 分布
ヒト血漿蛋白結合率は低く(20%以下)、高齢者及び腎機能障害患者の試験から得られたヒト血漿蛋白結合率も同様であった12)。
16.4 代謝
16.5 排泄
16.5.1 バレニクリンの腎排泄は主として糸球体濾過によるものであるが、有機カチオントランスポーターOCT-2を介した尿細管からの分泌排泄も一部寄与している14)。
16.5.2 健康成人男性(外国人)6例に14C-標識バレニクリン1mgを単回経口投与した時、投与148時間後までに投与放射能の87.1%及び0.9%が、それぞれ尿中及び糞中に排泄された。尿中に排泄された放射能のほとんどが未変化体(投与放射能の80.5%、尿中に排泄された放射能の91.6%)であったことから、経口投与されたバレニクリンの吸収率は高く、肝代謝をほとんど受けずに主として未変化体として尿中排泄される15)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
軽度の腎機能障害を有する被験者(クレアチニン・クリアランス(CLCR)推定値:50mL/分<CLCR<80mL/分)では、バレニクリンの薬物動態に対する腎機能障害の影響は認められなかった。中等度(CLCR推定値:30mL/分≦CLCR≦50mL/分)及び重度(CLCR推定値:CLCR<30mL/分)の腎機能障害を有する被験者では、腎機能が正常な被験者(CLCR推定値:CLCR>80mL/分)と比較してバレニクリンの全身曝露量がそれぞれ1.5倍及び2.1倍に増加した。また、週3回3時間の透析を行っている腎疾患を有する被験者では、バレニクリンの全身曝露量が2.7倍に増加した。なお、血液透析での除去率を検討した結果、血液透析は健康被験者における腎機能とほぼ同程度の排泄効果があると考えられた16)(外国人データ)。[7.6、9.2.1、9.2.2、9.8、10.2、13.1参照]
| 薬物動態パラメータ | 試験群 | 対照群 | 幾何平均値(LS平均) | 比a)(%) | 比の90%信頼区間 | |
| 試験群 | 対照群 | |||||
| AUC0-τb)(ng・h/mL) | 軽度 | 正常 | 58.68 | 55.55 | 105.64 | (79.35,140.65) |
| 中等度 | 84.38 | 151.92 | (114.11,202.26) | |||
| 重度 | 114.91 | 206.88 | (155.38,275.43) | |||
| 透析患者c) | 150.79 | 271.48 | (203.91,361.44) | |||
| Cmax(ng/mL) | 軽度 | 正常 | 3.67 | 3.99 | 91.92 | (70.33,120.15) |
| 中等度 | 4.83 | 120.98 | (92.56,158.13) | |||
| 重度 | 6.10 | 152.97 | (117.04,199.94) | |||
| 透析患者c) | 7.30 | 183.03 | (140.04,239.23) | |||
16.6.2 肝機能障害患者
バレニクリンはその大部分が未変化体として尿中に排泄され、ほとんど肝代謝を受けないことから、バレニクリンの薬物動態は肝障害の影響を受けないことが予測される。
16.6.3 高齢者
健康高齢男女喫煙者16例(65〜75歳)にバレニクリンを反復投与(1mg1日1回又は1日2回7日間)した時、バレニクリンの高齢喫煙者における薬物動態は、非高齢喫煙者と同様であった12)(外国人データ)。
16.6.4 小児
12〜17歳の喫煙者22例にバレニクリン0.5及び1mgを単回投与した時、バレニクリンの薬物動態はほぼ用量に比例し、全身曝露量及び腎クリアランスは、健康成人被験者と同様であった17)(外国人データ)。
16.7 薬物相互作用
16.7.1 In vitro試験
バレニクリンは肝ミクロソームによるチトクロームP450酵素(1A2、2A6、2B6、2C8、2C9、2C19、2D6、2E1及び3A4/5)の基質代謝を阻害しなかった(IC50>6,400ng/mL)。また、バレニクリンはヒト肝細胞のチトクロームP450酵素1A2及び3A4の活性を誘導しなかった。ヒト腎トランスポーター(hOCT-2、hOAT-1、hOAT-3、hOCTN-1又はhOCTN-2)を発現させたヒト胎児腎細胞への取り込みを検討した結果、バレニクリンはhOCT-2の基質であることが示され、シメチジン(1mM、OCT-2の阻害剤)によってバレニクリンの取り込みは部分的に阻害された18)19)。
16.7.2 臨床試験
(1)シメチジン
健康成人喫煙者12例にシメチジンを反復投与(300mg1日4回5日間)し、2日目にバレニクリン2mgを単回併用投与した時のバレニクリンの薬物動態は、バレニクリン単独投与時に比べて全身曝露量が約29%増加し(90%信頼区間:21.5%、36.9%)、投与48時間後までの腎クリアランスは約25%低下した20)(外国人データ)。[10.2参照]
注)本剤の承認用量は1回1mgまでである。
(2)その他の薬剤
16.7.3 他の禁煙補助薬(ニコチン代替療法)との併用
喫煙者22例に経皮吸収ニコチン製剤(21mg/日)とバレニクリン(1mg1日2回)を併用反復投与(ニコチン14日間反復貼付期間中3日目からバレニクリン反復投与)した時、ニコチンの薬物動態に対する影響はなかったが、14日目に測定した最高血圧の平均値に統計学的に有意な低下(平均2.6mmHg)が認められた。副作用は経皮吸収ニコチン製剤単独投与群17例中14例(82.4%)、併用投与群22例中17例(77.3%)に認められた。嘔気、頭痛、嘔吐、浮動性めまい、消化不良及び疲労は併用投与群で多く認められ、その発現率は経皮吸収ニコチン製剤単独投与群で嘔気7例(41.2%)、頭痛4例(23.5%)、嘔吐2例(11.8%)、浮動性めまい1例(5.9%)、消化不良1例(5.9%)及び疲労3例(17.6%)、併用投与群で嘔気14例(63.6%)、頭痛11例(50.0%)、嘔吐7例(31.8%)、浮動性めまい7例(31.8%)、消化不良5例(22.7%)及び疲労6例(27.3%)であったが、いずれの有害事象も安全性上の問題は認められなかった。なお、これらの有害事象は他の試験のバレニクリン単独投与でも認められている。また、本試験で検討したニコチン代替療法を含め、本剤を他の禁煙補助薬と併用した場合の安全性及び有効性に関する試験は行われていない24)(外国人データ)。[7.1参照]
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国内後期第II相用量反応試験
禁煙を希望する喫煙者を対象とした12週間投与の二重盲検比較試験において、主要評価項目の第9〜12週の4週間持続禁煙率は、バレニクリン1mg1日2回投与群で65.4%(85/130例)、プラセボ群で39.5%(51/129例)であり、バレニクリン1mg1日2回投与群はプラセボ群と比較して統計学的に有意に高かった。また、第9〜52週の持続禁煙率もプラセボ群と比較して優れていた。さらに、プラセボ群と比べて、離脱症状、タバコに対する切望感、喫煙から得られる満足感を軽減した。
| 薬剤名 | 第9〜12週の4週間持続禁煙率a) | 第9〜52週の持続禁煙率b) | ||||
| %(n/N) | オッズ比c) (95%信頼区間) | p値c) | %(n/N) | オッズ比c) (95%信頼区間) | p値c) | |
| バレニクリン 1mg1日2回 | 65.4(85/130) | 2.98 (1.78,4.99) | <0.0001 | 34.6(45/130) | 1.81 (1.04,3.17) | 0.0355 |
| バレニクリン 0.5mg1日2回 | 55.5(71/128) | 1.94 (1.17,3.22) | 0.0095 | 28.9(37/128) | 1.38 (0.78,2.46) | 0.2645 |
| バレニクリン 0.25mg1日2回 | 54.7(70/128) | 1.88 (1.14,3.12) | 0.0134 | 27.3(35/128) | 1.25 (0.70,2.23) | 0.4456 |
| プラセボ | 39.5(51/129) | − | − | 23.3(30/129) | − | − |
副作用の発現率は、バレニクリン0.25mg群で43.1%、0.5mg群で42.6%、1mg群で53.8%及びプラセボ群で28.6%であった。発現率5%以上の副作用は、上腹部痛、便秘、嘔気及び頭痛であった25)。
17.1.2 外国後期第II相用量反応試験
禁煙を希望する喫煙者を対象とした12週間投与の二重盲検比較試験において、主要評価項目の第9〜12週の4週間持続禁煙率は、バレニクリン1mg1日2回投与群ではプラセボ群と比較して統計学的に有意に高かった。また、第9〜52週の持続禁煙率もプラセボ群と比較して優れていた。
| 薬剤名 | 第9〜12週の4週間持続禁煙率a) | 第9〜52週の持続禁煙率b) | ||||
| %(n/N) | オッズ比c) (95%信頼区間) | p値c) | %(n/N) | オッズ比c) (95%信頼区間) | p値c) | |
| バレニクリン 1mg1日2回 | 55.0(71/129) | 10.23 (5.24,19.98) | <0.0001 | 25.6(33/129) | 9.02 (3.33,24.43) | <0.0001 |
| バレニクリン 0.5mg1日2回 | 41.1(53/129) | 5.34 (2.75,10.36) | <0.0001 | 18.6(24/129) | 5.76 (2.09,15.89) | 0.0001 |
| プラセボ | 12.4(15/121) | − | − | 4.1(5/121) | − | − |
国内外の後期第II相用量反応試験において、第9〜12週の4週間持続禁煙率及び第9〜52週持続禁煙率はバレニクリンの用量に依存して上昇した。
17.1.3 国内再投与試験
バレニクリンを再投与した際の安全性を評価する目的で、国内後期第II相用量反応試験の第9〜12週に持続禁煙できなかった喫煙者42例に本剤を12週間再投与したところ、安全性に問題がないことが示された。報告された有害事象とその発現頻度及び重症度は、国内後期第II相用量反応試験と同様であった。また、本剤の再投与により持続禁煙に成功した被験者が認められた。
副作用の発現率は、バレニクリン0.25mg群で35.7%、0.5mg群で36.4%、1mg群で38.5%、プラセボ群で25.0%であった。いずれかのバレニクリン群で発現率20%以上の副作用は、嘔気であった28)。
副作用の発現率は、バレニクリン0.25mg群で35.7%、0.5mg群で36.4%、1mg群で38.5%、プラセボ群で25.0%であった。いずれかのバレニクリン群で発現率20%以上の副作用は、嘔気であった28)。
17.1.4 外国第III相比較検証試験(2試験)
12週間投与のプラセボを対照とした2つの二重盲検比較試験において、主要評価項目の第9〜12週の4週間持続禁煙率は、バレニクリン1mg1日2回投与群でプラセボ群と比較して統計学的に有意に高かった。
| 試験 | 薬剤名 | 第9〜12週の4週間持続禁煙率a) | 第9〜52週の持続禁煙率b) | ||||
| %(n/N) | オッズ比c) (95%信頼区間) | p値c) | %(n/N) | オッズ比c) (95%信頼区間) | p値c) | ||
| 試験1 | バレニクリン 1mg1日2回 | 44.4(155/349) | 3.91 (2.74,5.59) | <0.0001 | 22.1(77/349) | 3.13 (1.97,4.97) | <0.0001 |
| プラセボ | 17.7(61/344) | − | − | 8.4(29/344) | − | − | |
| 試験2 | バレニクリン 1mg1日2回 | 44.0(151/343) | 3.85 (2.69,5.50) | <0.0001 | 23.0(79/343) | 2.66 (1.72,4.11) | <0.0001 |
| プラセボ | 17.7(60/340) | − | − | 10.3(35/340) | − | − | |
17.1.5 外国第III相禁煙維持療法試験
バレニクリン1mg1日2回を非盲検下で12週間投与し、第12週までに禁煙できた患者にバレニクリン1mg1日2回又はプラセボを二重盲検下で12週間追加投与し、禁煙維持に対する本剤の有効性及び安全性を評価した。主要評価項目の第13〜24週の持続禁煙率は、バレニクリン1mg1日2回投与群で70.6%(425/602例)であり、プラセボ群の49.8%(301/604例)と比較して統計学的に有意に高かった。
| 薬剤名 | 第13〜24週持続禁煙率a) | 第13〜52週持続禁煙率b) | ||||
| %(n/N) | オッズ比c) (95%信頼区間) | p値c) | %(n/N) | オッズ比c) (95%信頼区間) | p値c) | |
| バレニクリン 1mg1日2回 | 70.6(425/602) | 2.47 (1.95,3.15) | <0.0001 | 44.0(265/602) | 1.35 (1.07,1.70) | 0.0126 |
| プラセボ | 49.8(301/604) | − | − | 37.1(224/604) | − | − |
17.1.6 外国臨床試験において、最大3%の患者でバレニクリンの投与終了によって易刺激性、喫煙衝動、抑うつ、あるいは不眠症の増強が認められた。
18. 薬効薬理
18.1 作用機序
バレニクリンは、α4β2ニコチン受容体に対して高い結合親和性をもつ、ニコチン受容体の部分作動薬である。バレニクリンが脳内のα4β2ニコチン受容体に結合すると、ニコチンを遮断して喫煙による満足感を抑制する(拮抗作用)。同時に、ニコチンの作用で放出されるよりも少量のドパミンを放出させ、禁煙に伴う離脱症状やタバコに対する切望感を軽減する(刺激作用)。
18.2 ニコチン受容体結合能
18.3 ニコチン受容体部分作動薬作用
18.4 ニコチン摂取の抑制作用
バレニクリンはニコチン依存ラットにおけるニコチン自己摂取行動を抑制した39)。
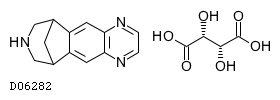
19. 有効成分に関する理化学的知見
19.1. バレニクリン酒石酸塩
| 一般的名称 | バレニクリン酒石酸塩 |
|---|---|
| 一般的名称(欧名) | Varenicline Tartrate |
| 化学名 | 7,8,9,10-Tetrahydro-6H-6,10-methanoazepino[4,5-g]quinoxaline mono[(2R,3R)-tartrate] |
| 分子式 | C13H13N3・C4H6O6 |
| 分子量 | 361.35 |
| 物理化学的性状 | バレニクリン酒石酸塩は、白色〜微黄色の結晶性の粉末であり、水に溶けやすく、N,N-ジメチルアセトアミドに溶けにくく、エタノール(99.5)にほとんど溶けない。 |
| KEGG DRUG |
|
22. 包装
<チャンピックススタート用パック>
(0.5mg×11錠、1mg×14錠)×1パック
<チャンピックス錠0.5mg>
28錠[14錠(PTP)×2]
<チャンピックス錠1mg>
28錠[14錠(PTP)×2]
84錠[14錠(PTP)×6]
23. 主要文献
- Kawakami N,et al., Addict Behav., 24 (2), 155-166, (1999) »PubMed
- たか橋 三郎ほか訳, DSM-IV-TR 精神疾患の診断・統計マニュアル 新訂版, 259, (2004), (医学書院)
- Rigotti NA,et al., Circulation., 121 (2), 221-229, (2010) »PubMed
- 社内資料:心血管イベントに関する安全性メタ解析
- Ware JH,et al., Am J Ther., 20 (3), 235-246, (2013) »PubMed
- 社内資料:自己摂取強化作用(2008年1月25日承認、CTD2.6.2.3(1))
- 社内資料:薬物乱用性に関する臨床薬理試験(2008年1月25日承認、CTD2.7.4.5.6)
- 社内資料:ラットがん原性試験(2008年1月25日承認、CTD2.6.6.5(3))
- 社内資料:健康成人における薬物動態(単回投与)(2008年1月25日承認、CTD2.5.3.1.1.1)
- 社内資料:健康成人における薬物動態(反復投与)(2008年1月25日承認、CTD2.5.3.1.1.1)
- 社内資料:健康成人における薬物動態(食事の影響)(2008年1月25日承認、CTD2.7.1.2.1.2)
- Burstein AH,et al., J Clin Pharmacol., 46 (11), 1234-1240, (2006) »PubMed
- 社内資料:代謝物の検討(2008年1月25日承認、CTD2.6.4.5)
- 社内資料:排泄の検討(2008年1月25日承認、CTD2.6.4.5、2.6.4.6)
- 社内資料:健康喫煙者での代謝・排泄作用(2008年1月25日承認、CTD2.7.2.2.2.1.2.4)
- 社内資料:腎機能障害患者での薬物動態(2008年1月25日承認、CTD2.7.2.3.2.4)
- 社内資料:青少年における薬物動態(2008年1月25日承認、CTD2.7.2.3.2.2)
- 社内資料:酵素阻害の検討(2008年1月25日承認、CTD2.6.4.5(3)、2.6.5.12)
- 社内資料:腎クリアランス機構の検討(2008年1月25日承認、CTD2.6.4.6(4)、2.6.5.13)
- 社内資料:シメチジンとの薬物相互作用(2008年1月25日承認、CTD2.7.2.2.4.3.1)
- 社内資料:メトホルミンとの薬物相互作用(2008年1月25日承認、CTD2.7.2.2.4.3.2)
- 社内資料:ジゴキシンとの薬物相互作用(2008年1月25日承認、CTD2.7.2.2.4.1.1)
- Burstein AH,et al., J Clin Pharmacol., 47 (11), 1421-1429, (2007) »PubMed
- 社内資料:ニコチン置換療法との薬物相互作用(2008年1月25日承認、CTD2.7.4.5.3.3.2)
- Nakamura M,et al., Clin Ther., 29 (6), 1040-1056, (2007) »PubMed
- Oncken C,et al., Arch Intern Med., 166 (15), 1571-1577, (2006) »PubMed
- 社内資料:外国後期第II相用量反応試験(2008年1月25日承認、CTD2.7.3.3.2.1.6)
- 社内資料:国内再投与試験(2008年1月25日承認、CTD2.7.4.1.1.3.1(2))
- Gonzales D,et al., JAMA., 296 (1), 47-55, (2006) »PubMed
- Jorenby DE,et al., JAMA., 296 (1), 56-63, (2006) »PubMed
- 社内資料:外国第III相比較検証試験(2試験)(2008年1月25日承認、CTD2.7.3.2.3.1)
- Tonstad S,et al., JAMA., 296 (1), 64-71, (2006) »PubMed
- 社内資料:外国第III相禁煙維持療法試験(2008年1月25日承認、CTD2.7.3.3.2.2.3)
- 社内資料:ニコチン受容体結合作用(2008年1月25日承認、CTD2.6.2.2(1))
- 社内資料:種々の受容体結合作用(2008年1月25日承認、CTD2.6.2.2(1))
- 社内資料:ニコチン受容体の部分作動薬作用(2008年1月25日承認、CTD2.6.2.2(2))
- 社内資料:ニコチン受容体の部分作動薬作用(ドパミン遊離促進作用)(2008年1月25日承認、CTD2.6.2.2(2))
- 社内資料:ニコチン受容体の部分作動薬作用(ドパミン遊離抑制作用)(2008年1月25日承認、CTD2.6.2.2(2))
- 社内資料:ニコチン自己摂取抑制作用(2008年1月25日承認、CTD2.6.2.2(3))
24. 文献請求先及び問い合わせ先
文献請求先
ファイザー株式会社
Pfizer Connect/メディカル・インフォメーション
〒151-8589
東京都渋谷区代々木3-22-7
電話:0120-664-467
製品情報問い合わせ先
ファイザー株式会社
Pfizer Connect/メディカル・インフォメーション
〒151-8589
東京都渋谷区代々木3-22-7
電話:0120-664-467
25. 保険給付上の注意
25.1 本製剤の薬剤料注)については、ニコチン依存症管理料の算定に伴って処方された場合に限り算定できることとする。また、処方箋による投薬の場合においては、処方箋の「備考」欄に「ニコチン依存症管理料の算定に伴う処方である。」と記載すること。
注)「用法及び用量に関連する注意」7.3又は7.4の処方に係る投与については、算定することは出来ない。
25.2 25.1にかかわらず、ニコチン依存症管理料を算定する禁煙治療を行っている患者が、何らかの理由により入院治療を要することとなった場合、ニコチン依存症管理料の施設基準を届け出ている保険医療機関に入院し、患者本人の強い禁煙意志に基づき禁煙治療を継続した場合に限り、当該禁煙治療に要した本剤の薬剤料を、入院している保険医療機関において算定して差し支えない。
当該薬剤料の算定に当たっては、外来で実施されていた禁煙治療の内容を十分に踏まえ、継続して計画的な禁煙指導を行うために本剤が処方された場合に算定が認められるものであること。
また、診療報酬請求の際には、診療報酬明細書の摘要欄に、「外来にてニコチン依存症管理料の算定患者に対し処方」と記載すること。
なお、入院の期間は、ニコチン依存症管理料の算定期間である12週間には含めないものとし、また、当該入院中の処方については、ニコチン依存症管理料を算定できる治療回数である5回には含めない。
当該薬剤料の算定に当たっては、外来で実施されていた禁煙治療の内容を十分に踏まえ、継続して計画的な禁煙指導を行うために本剤が処方された場合に算定が認められるものであること。
また、診療報酬請求の際には、診療報酬明細書の摘要欄に、「外来にてニコチン依存症管理料の算定患者に対し処方」と記載すること。
なお、入院の期間は、ニコチン依存症管理料の算定期間である12週間には含めないものとし、また、当該入院中の処方については、ニコチン依存症管理料を算定できる治療回数である5回には含めない。
26. 製造販売業者等
26.1 製造販売元
ファイザー株式会社
東京都渋谷区代々木3-22-7