医薬品情報
| 総称名 | シーエルセントリ |
|---|---|
| 一般名 | マラビロク |
| 欧文一般名 | Maraviroc |
| 製剤名 | マラビロク錠 |
| 薬効分類名 | 抗ウイルス化学療法剤(CCR5阻害剤) |
| 薬効分類番号 | 6250 |
| ATCコード | J05AX09 |
| KEGG DRUG |
D06670
マラビロク
|
| KEGG DGROUP |
DG03107
抗HIV薬
|
| JAPIC | 添付文書(PDF) |
添付文書情報2026年3月 改訂(第6版)

|
2.禁忌 4.効能または効果 5.効能又は効果に関連する注意 6.用法及び用量 7.用法及び用量に関連する注意 8.重要な基本的注意 9.特定の背景を有する患者に関する注意 10.相互作用 11.副作用 15.その他の注意 16.薬物動態 17.臨床成績 18.薬効薬理 19.有効成分に関する理化学的知見 22.包装 23.主要文献 24.文献請求先及び問い合わせ先 26.製造販売業者等 |
商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| シーエルセントリ錠150mg | CELSENTRI Tablets 150mg | ヴィーブヘルスケア | 6250034F1025 | 1148.2円/錠 | 劇薬, 処方箋医薬品 |
2. 禁忌
次の患者には投与しないこと
本剤の成分に対し過敏症の既往歴のある患者
4. 効能または効果
CCR5指向性HIV-1感染症
5. 効能または効果に関連する注意
5.2 CXCR4指向性ヒト免疫不全ウイルス(HIV)-1感染患者、CCR5/CXCR4二重又は混合指向性HIV-1感染患者には、投与しないこと。なお、急性期及び無症候期の患者では主にCCR5指向性ウイルスが検出されるが、進行したHIV-1感染症ではCXCR4指向性及び二重/混合指向性ウイルスが検出される患者の割合が増加することが知られている。
6. 用法及び用量
通常、成人にはマラビロクとして1回300mgを1日2回経口投与する。なお、投与に際しては必ず他の抗HIV薬を併用し、併用薬に応じて適宜増減すること。本剤は、食事の有無にかかわらず投与できる。
7. 用法及び用量に関連する注意
7.1 CYP3A阻害剤又はCYP3A誘導剤と併用する場合には、下表を参照し、本剤の用量調整を行うこと。[10.、10.2、16.4.1参照]
| 併用薬 | 本剤の用量 |
| 以下の強力なCYP3A阻害剤(CYP3A誘導剤の有無を問わない): ・プロテアーゼ阻害剤(tipranavir+リトナビルを除く) ・テラプレビル ・デラビルジン ・イトラコナゾール、ケトコナゾール、クラリスロマイシン ・その他の強力なCYP3A阻害剤(nefazodone、テリスロマイシン等) | 150mg1日2回 |
| tipranavir+リトナビル、ネビラピン、ラルテグラビル、あらゆるヌクレオシド系逆転写酵素阻害剤(NRTI)及びenfuvirtide等のその他の併用薬 | 300mg1日2回 |
| 以下の強力なCYP3A誘導剤(強力なCYP3A阻害剤の併用なし): ・エファビレンツ、エトラビリン ・リファンピシン ・カルバマゼピン、フェノバルビタール、フェニトイン 等 | 600mg1日2回 |
7.2 1回300mg、1日2回を上回る用法・用量での有効性及び安全性は確立していない(投与経験がない)。
7.3 腎機能障害(クレアチニンクリアランス(Ccr)<80mL/min)があり、強力なCYP3A4阻害剤を投与している患者では、腎機能の低下に応じて、次の投与間隔及び投与量を目安に投与すること。ただし、これらの投与間隔の調節に対する有効性及び安全性は確立されていないため、患者の臨床症状等を十分に観察すること(外国人データ)。[9.2.1、9.2.2、16.6.1参照]
| 併用薬 | Ccr<80mL/min |
| 強力なCYP3A4阻害剤を併用しない時又はtipranavir+リトナビル併用時 | 投与間隔の調節は必要ない(300mgを12時間毎) |
| ホスアンプレナビル+リトナビル併用時 | 150mgを12時間毎 |
| 以下の強力なCYP3A4阻害剤の併用時: ・サキナビル+リトナビル併用時 ・ロピナビル・リトナビル、ダルナビル+リトナビル、アタザナビル+リトナビル、ケトコナゾール等 | 150mgを24時間毎 |
8. 重要な基本的注意
8.1 健康成人を対象とした臨床試験において、本剤によると疑われるアレルギー症状を伴う肝障害が1例報告されている。また、治療歴の有無に関わらずHIV感染患者を対象とした臨床試験において、肝機能検査異常の増加や肝障害が報告されたが、グレード3及び4注)の肝機能検査異常の増加は認められなかった。本剤投与後に肝炎あるいは全身性アレルギー症状(そう痒性皮疹、好酸球増加、IgE上昇等)が認められた場合には、投与を中止するなど適切な処置を行うこと。
注)エイズ臨床試験グループ(ACTG)分類
8.2 本剤の使用に際しては、国内外のガイドライン等の最新の情報を参考に、患者又は患者に代わる適切な者に、次の事項についてよく説明し同意を得た後、使用すること。
・本剤はHIV感染症の根治療法薬ではないことから、日和見感染症を含むHIV感染症の進展に伴う疾病を発症し続ける可能性があるので、本剤投与開始後の身体状況の変化については全て担当医に報告すること。
・担当医の指示なしに用量を変更したり、服用を中止したりしないこと。
・本剤の長期投与による影響については現在のところ不明であること。
・本剤は併用薬剤と相互作用を起こすことがあるため、服用中の全ての薬剤を担当医に報告すること。また、本剤で治療中に新たに他の薬剤を服用する場合には、事前に担当医師に相談すること。
8.3 ウイルスの指向性検査は、有用性が確立された高感度な方法により行うこと。ウイルスの指向性は、患者の治療歴又は保存検体の検査から推測することはできないため、最新の検体で指向性検査を実施すること。[5.1、8.4参照]
8.5 ウイルス学的効果が認められなかった場合は、指向性検査の結果にかかわらず本剤の継続投与は推奨されない。
8.6 本剤を含む抗HIV薬の多剤併用療法を行った患者で、免疫再構築症候群が報告されている。投与開始後、免疫機能が回復し、症候性のみならず無症候性日和見感染(マイコバクテリウムアビウムコンプレックス、サイトメガロウイルス、ニューモシスチス等によるもの)等に対する炎症反応が発現することがある。また、免疫機能の回復に伴い自己免疫疾患(甲状腺機能亢進症、多発性筋炎、ギラン・バレー症候群、ブドウ膜炎等)が発現するとの報告があるので、これらの症状を評価し、必要時には適切な治療を考慮すること。
8.7 本剤は、免疫細胞のCCR5コレセプターを阻害することから、感染症発症の危険性を増大させる可能性がある。本剤投与中は、感染症の徴候について十分な観察を行い、必要に応じて適切な処置を行うこと。
8.8 本剤投与に伴う悪性腫瘍の増加は認められていないが、免疫機構に影響を及ぼす可能性があるため、悪性腫瘍発症の危険性が増大するおそれがある。
8.9 めまい等があらわれることがあるので、高所作業、自動車の運転等危険を伴う機械を操作する際には注意させること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 重篤な心疾患又はその既往歴のある患者
心筋虚血等をおこすおそれがある。[11.1.1参照]
9.1.2 B型・C型肝炎の患者
9.1.3 起立性低血圧の既往歴のある患者
起立性低血圧をおこすおそれがある。
9.1.4 降圧作用を有する併用薬の投与を受けている患者[10.2参照]
9.2 腎機能障害患者
9.2.1 重度の腎機能障害のある患者
9.2.2 腎機能障害(Ccr<80mL/min)のある患者(重度の腎機能障害のある患者を除く)
9.3 肝機能障害患者
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。
9.6 授乳婦
授乳を避けさせること。動物実験(ラット)で乳汁への移行が報告されている。また、HIV感染女性患者は、乳児のHIV感染を避けるため、乳児に母乳を与えないことが望ましい。
9.7 小児等
小児等を対象とした臨床試験は実施していない。
9.8 高齢者
一般に生理機能が低下していることが多い。
10. 相互作用
相互作用序文
薬物代謝酵素用語
CYP3A4
10.2 併用注意
| HIVプロテアーゼ阻害剤 アタザナビル硫酸塩 アタザナビル硫酸塩+リトナビル ロピナビル・リトナビル サキナビル+リトナビル ダルナビルエタノール付加物+リトナビル ネルフィナビルメシル酸塩 ホスアンプレナビルカルシウム水和物+リトナビル [7.1、16.7.2参照] | 本剤の血中濃度が上昇するおそれがあるので、本剤の用量を150mg1日2回に減量すること。 | これらのプロテアーゼ阻害剤はCYP3A4の代謝活性を阻害するため、本剤の血中濃度が上昇するおそれがある。 |
| HIVプロテアーゼ阻害剤+非ヌクレオシド系逆転写酵素阻害剤(NNRTI) HIVプロテアーゼ阻害剤(tipranavir+リトナビルを除く)+エファビレンツ又はエトラビリン [7.1、16.7.2参照] | 本剤の血中濃度が上昇するおそれがあるので、本剤の用量を150mg1日2回に減量すること。 | これらのプロテアーゼ阻害剤はCYP3A4の代謝活性を阻害するため、本剤の血中濃度が上昇するおそれがある。 |
| HIVプロテアーゼ阻害剤(tipranavir+リトナビルを除く)+リファブチン [7.1参照] | 本剤の血中濃度が上昇するおそれがあるので、本剤の用量を150mg1日2回に減量すること。 | これらのプロテアーゼ阻害剤はCYP3A4の代謝活性を阻害するため、本剤の血中濃度が上昇するおそれがある。 |
| NNRTI デラビルジンメシル酸塩 [7.1参照] | 本剤の血中濃度が上昇するおそれがあるので、本剤の用量を150mg1日2回に減量すること。 | これらの薬剤はCYP3A4の代謝活性を阻害するため、本剤の血中濃度が上昇するおそれがある。 |
| 抗真菌剤 イトラコナゾール ケトコナゾール [7.1、16.7.2参照] | 本剤の血中濃度が上昇するおそれがあるので、本剤の用量を150mg1日2回に減量すること。 | これらの薬剤はCYP3A4の代謝活性を阻害するため、本剤の血中濃度が上昇するおそれがある。 |
| 抗菌剤 クラリスロマイシン テリスロマイシン [7.1参照] | 本剤の血中濃度が上昇するおそれがあるので、本剤の用量を150mg1日2回に減量すること。 | これらの薬剤はCYP3A4の代謝活性を阻害するため、本剤の血中濃度が上昇するおそれがある。 |
| テラプレビル [7.1、16.7.2参照] | 本剤の血中濃度が上昇するおそれがあるので、本剤の用量を150mg1日2回に減量すること。 | これらの薬剤はCYP3A4の代謝活性を阻害するため、本剤の血中濃度が上昇するおそれがある。 |
| nefazodone | 本剤の血中濃度が上昇するおそれがあるので、本剤の用量を150mg1日2回に減量すること。 | これらの薬剤はCYP3A4の代謝活性を阻害するため、本剤の血中濃度が上昇するおそれがある。 |
| NNRTI エファビレンツ エトラビリン [7.1、16.7.2参照] | 本剤の血中濃度が低下するおそれがあるので、強力なCYP3A4阻害剤を併用せずにこれらの薬剤を併用投与する場合、本剤の用量を600mg1日2回に増量すること。 | これらの薬剤はCYP3A4の代謝活性を誘導するため、本剤の血中濃度が低下するおそれがある。 |
| 抗菌剤 リファンピシン [7.1、16.7.2参照] | 本剤の血中濃度が低下するおそれがあるので、強力なCYP3A4阻害剤を併用せずにこれらの薬剤を併用投与する場合、本剤の用量を600mg1日2回に増量すること。 | これらの薬剤はCYP3A4の代謝活性を誘導するため、本剤の血中濃度が低下するおそれがある。 |
| カルバマゼピン フェノバルビタール フェニトイン [7.1参照] | 本剤の血中濃度が低下するおそれがあるので、強力なCYP3A4阻害剤を併用せずにこれらの薬剤を併用投与する場合、本剤の用量を600mg1日2回に増量すること。 | これらの薬剤はCYP3A4の代謝活性を誘導するため、本剤の血中濃度が低下するおそれがある。 |
| リファンピシン+エファビレンツ [7.1、16.7.2参照] | 本剤の血中濃度が著しく低下して至適水準を下回り、ウイルス学的効果の消失や本剤に対する耐性が生じる可能性があるので、本剤とこれらの薬剤の併用は推奨されない。 | これらの薬剤等はCYP3A4の代謝活性を誘導するため、本剤の血中濃度が著しく低下するおそれがある。 |
| セイヨウオトギリソウ(St.John's Wort、セント・ジョーンズ・ワート)含有食品 | 本剤の血中濃度が著しく低下して至適水準を下回り、ウイルス学的効果の消失や本剤に対する耐性が生じる可能性があるので、本剤投与時はセイヨウオトギリソウ含有食品を摂取しないように注意すること。 | これらの薬剤等はCYP3A4の代謝活性を誘導するため、本剤の血中濃度が著しく低下するおそれがある。 |
| 降圧作用を有する薬剤 アムロジピン オルメサルタン ビソプロロール等 [9.1.4参照] | 本剤の血中濃度の上昇に相関して、起立性低血圧が発現することが確認されている。本剤と降圧作用を有する薬剤とを併用した場合に起立性低血圧が発現することを示す試験はないものの、降圧作用を有する薬剤を併用中の患者は、起立性低血圧及び低血圧に関連する症状の発現に十分注意する必要がある。 | 機序不明 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 心筋虚血(0.5%未満)[9.1.1参照]
11.1.3 肺炎、食道カンジダ症(いずれも0.5%未満)
11.1.4 胆管癌、骨転移、肝転移、腹膜転移(いずれも0.5%未満)
11.1.5 汎血球減少症、好中球減少症、リンパ節症(いずれも0.5%未満)
11.1.6 幻覚(0.5%未満)
11.1.7 脳卒中、意識消失、てんかん、小発作てんかん、痙攣、顔面神経麻痺、多発ニューロパシー、反射消失(いずれも0.5%未満)
11.1.8 白内障(0.5%未満)
11.1.9 呼吸窮迫、気管支痙攣(いずれも0.5%未満)
11.1.10 膵炎、直腸出血(いずれも0.5%未満)
11.1.11 筋炎(0.5%未満)
11.1.12 腎不全、多尿(いずれも0.5%未満)
11.1.13 皮膚粘膜眼症候群(Stevens-Johnson症候群)(0.5%未満)
11.2 その他の副作用
| 2%以上 | 2%未満 | |
| 血液 | 貧血 | ヘマトクリット減少、ヘモグロビン減少、好中球数減少、白血球数減少、血小板数減少 |
| 感染症及び寄生虫症 | 鼻咽頭炎、耳感染、真菌感染、感染性筋炎、インフルエンザ、ウイルス感染 | |
| 代謝及び栄養障害 | 高トリグリセリド血症、高血糖、食欲亢進、食欲減退、インスリン抵抗性糖尿病、多飲症 | |
| 精神障害 | 不眠症 | 異常な夢、うつ病、感情障害、気分循環性障害、失見当識、多幸気分、リビドー減退、気分変動 |
| 神経系障害 | 浮動性めまい、味覚異常、頭痛 | 錯感覚、傾眠、感覚鈍麻、末梢性ニューロパシー、失神、精神運動亢進、レストレスレッグス症候群、振戦、味覚消失、健忘、記憶障害、異常感覚、副鼻腔炎に伴う頭痛、三叉神経痛 |
| 眼障害 | 眼刺激、眼乾燥、眼痛、弱視、アレルギー性結膜炎 | |
| 耳及び迷路障害 | 耳痛、乗物酔い、耳漏、鼓膜充血 | |
| 心臓障害 | 第一度房室ブロック、徐脈、頻脈、動悸 | |
| 血管障害 | ほてり、レイノー現象、起立性低血圧 | |
| 呼吸器、胸郭及び縦隔障害 | 咳嗽 | 鼻閉、鼻乾燥、季節性鼻炎、呼吸困難、発声障害、肺気腫、肺障害、咽頭紅斑、咽喉頭不快感、咽喉頭疼痛、咽喉絞扼感、低音性連続性ラ音、上気道うっ血 |
| 胃腸障害 | 便秘、腹痛、消化不良、悪心、鼓腸、嘔吐、下痢 | 口の錯感覚、口の感覚鈍麻、口唇水疱、口腔内潰瘍形成、口唇のひび割れ、舌痛、歯痛、嚥下障害、おくび、レッチング、腹部膨満、胃食道逆流性疾患、腹部不快感、消化器痛、白色便、異常便、排便痛 |
| 肝胆道系障害 | 肝脾腫大、黄疸 | |
| 皮膚及び皮下組織障害 | 発疹 | 脱毛症、紅斑、体脂肪の再分布/蓄積、ざ瘡、冷汗、湿疹、過角化、爪の障害、爪変色、皮膚灼熱感、皮膚剥脱、皮膚刺激、そう痒症、毛包炎 |
| 筋骨格系及び結合組織障害 | 背部痛、頚部痛、筋痙縮、四肢痛、筋痛、肋軟骨炎、鼡径部腫瘤、筋緊張、筋骨格痛、ミオパシー | |
| 腎及び尿路障害 | 夜間頻尿、尿失禁、蛋白尿、着色尿、血尿 | |
| 生殖系及び乳房障害 | 勃起不全、良性前立腺肥大症、乳房腫瘤、乳房圧痛、不正子宮出血、乳頭痛、骨盤痛 | |
| 全身障害及び投与局所様態 | 疲労 | 無力症、異常感、胸部不快感、胸痛、易刺激性、口渇、脂肪織増加、全身性浮腫、炎症、インフルエンザ様疾患、薬物不耐性、注射部位反応、注射部位硬結、注射部位疼痛 |
| 臨床検査 | ALT増加、AST増加、γGTP増加、血中クレアチンホスホキナーゼ増加、血中トリグリセリド増加、血中コレステロール増加、血中クレアチニン増加、血中鉄減少、血中カリウム減少、血中カリウム増加、ウイルス負荷増加、心電図QT延長、体温上昇、体重増加、体重減少 | |
| 傷害、中毒及び処置合併症 | 転倒、筋損傷、肋骨骨折 |
15. その他の注意
16. 薬物動態
16.1 血中濃度
16.1.1 単回経口投与
健康成人男性12例に本剤300mgを空腹時単回経口投与した時、マラビロクは投与後1.5〜5.0時間(中央値では3.0時間)に最高血漿中濃度(Cmax)に到達した。Cmax及び血漿中濃度−時間曲線下面積(AUC0-inf)の幾何平均値(変数係数%)はそれぞれ736ng/mL(42%)及び2763ng・h/mL(29%)であり、終末相の消失半減期(t1/2)の算術平均値(変数係数%)は13.0時間(23%)であった。
健康成人を対象に本剤300mgを単回経口投与した時、マラビロクは投与後0.5〜4時間(中央値では2時間)でCmaxに到達した3)。
健康成人を対象にマラビロク1〜1200mg注)を単回経口投与した時、マラビロクの薬物動態は投与量に比例しなかった4)(外国人データ)。
16.1.2 反復経口投与
健康成人及びHIV感染患者にマラビロクを投与した時の定常状態の薬物動態パラメータを表1に示す5)(外国人データ)。
| マラビロクの用量 | 例数 | Cmax(ng/mL) | AUC12(ng・h/mL) | Cmin(ng/mL) | |
| 健康成人 (第I相) | 300mg 1日2回 | 64 | 888 | 2908 | 43.1 |
| 無症候性HIV感染患者 (第IIa相) | 300mg 1日2回 | 8 | 618 | 2550 | 33.6 |
| 治療歴のあるHIV感染患者 (第III相)注1) | 300mg 1日2回 | 94 | 266 | 1513 | 37.2 |
| 150mg 1日2回 (CYP3A4阻害剤併用) | 375 | 332 | 2463 | 101 |
16.2 吸収
16.2.1 食事の影響
16.2.2 バイオアベイラビリティ
16.3 分布
16.3.1 分布容積
健康成人にマラビロク100mg注)を投与した時の分布容積は約194Lであった7)(外国人データ)。
16.3.2 血漿蛋白結合率
16.3.3 結合蛋白
In vitroで、マラビロクはアルブミン及びα1酸性糖蛋白と中等度の親和性を示す9)。
16.4 代謝
16.4.1 主な代謝酵素
16.4.2 In vivo試験
16.5 排泄
健康成人を対象にマラビロクを経口投与した時、定常状態におけるマラビロクの終末相の半減期は、14〜18時間であった。14C-マラビロク300mgを単回投与したマスバランス試験において、投与後168時間で放射能の約20%が尿中に回収され、76%が糞便中に回収された。尿中及び糞便中へは主として未変化体として排泄され、それぞれ投与量の8%及び25%(平均値)であった。その他は代謝物として排泄された7)(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
マラビロクの腎クリアランスは、CYP3A4を阻害する薬剤の非併用時では総クリアランスの約23%であるが、併用時では約70%を占める可能性がある。
腎機能障害患者における薬物動態のシミュレーション検討結果から、強力なCYP3A4阻害剤との併用時には、マラビロクの血中濃度が増加するため、投与量を減量する必要がある。
腎機能障害患者における薬物動態のシミュレーション検討結果から、強力なCYP3A4阻害剤との併用時には、マラビロクの血中濃度が増加するため、投与量を減量する必要がある。
(1)腎機能正常被験者6例、重度(Ccr<30mL/min)腎機能障害患者6例、及び週3回透析を行っている患者6例にマラビロク300mg単回投与を行った。AUCinf(変動係数%)は、腎機能正常被験者1348.4ng・h/mL(61%)、重度腎機能障害患者4367.7ng・h/mL(52%)、透析患者(透析後投与時)2677.4ng・h/mL(40%)、透析患者(透析前投与時)2805.5ng・h/mL(45%)であった。Cmax(変動係数%)はそれぞれ、335.6ng/mL(87%)、801.2ng/mL(56%)、576.7ng/mL(51%)、478.5ng/mL(38%)であった。なお、マラビロクの透析クリアランス(変動係数%)は36.4mL/min(33%)であった(外国人データ)。
(2)腎機能正常被験者6例にマラビロク150mg(12時間毎)とサキナビル1000mg+リトナビル100mg BIDの併用、軽度(Ccr>50〜≦80mL/min)腎機能障害患者6例にマラビロク150mg(24時間毎)とサキナビル1000mg+リトナビル100mg BIDの併用、中等度(Ccr≧30〜≦50mL/min)腎機能障害患者6例にマラビロク150mg(48時間毎注))とサキナビル1000mg+リトナビル100mg BIDの併用にて7日間経口投与をした時、腎機能正常被験者と比べて軽度腎機能障害患者ではAUCtau、Cmaxはそれぞれ52%、21%上昇し、Cminは43%低下した。また、中等度腎機能障害患者ではAUCtauは16%上昇し、Cmax及びCminはそれぞれ29%、85%低下した。
したがって、腎機能障害があり、強力なCYP3A4阻害剤を投与している患者では、マラビロクの投与量を150mg QDに調整する必要がある。なお、投与間隔を24時間以上とした場合は、投与後24〜48時間のマラビロクの曝露が不十分になる可能性がある(外国人データ)。[7.3、9.2.1、9.2.2参照]
したがって、腎機能障害があり、強力なCYP3A4阻害剤を投与している患者では、マラビロクの投与量を150mg QDに調整する必要がある。なお、投与間隔を24時間以上とした場合は、投与後24〜48時間のマラビロクの曝露が不十分になる可能性がある(外国人データ)。[7.3、9.2.1、9.2.2参照]
16.6.2 肝機能障害患者
マラビロクは主に肝臓で代謝され消失する。軽度(Child-Pugh分類A:8例)又は中等度(Child-Pugh分類B:8例)の肝機能障害を有する患者にマラビロク300mgを単回投与した時のマラビロクの薬物動態が検討されている。肝機能の正常な被験者(8例)と比較して軽度の肝機能障害患者のCmax及びAUC(平均値)はそれぞれ11%及び25%、中等度の肝機能障害患者ではそれぞれ32%及び46%高い値を示した11)。重度の肝機能障害を有する患者の薬物動態は検討されていない(外国人データ)。
16.6.3 小児等
小児患者における本剤の薬物動態は確立されていない(外国人データ)。
16.6.4 年齢
16.6.5 性別
臨床第I相及び第IIa相試験データを用いた母集団薬物動態解析の結果、性別(女性:96例、全集団の23.2%)はマラビロクの血中濃度には影響を及ぼさないことが示されている12)。性別による用量調節は不要である(外国人データ)。
16.6.6 人種
16.7 薬物相互作用
16.7.1 In vitro試験
(1)分布・排泄に関わるトランスポーター
In vitroにおいてマラビロクはP糖蛋白質(P-gp)及びOATP1B1の基質であり、P-gpを阻害する(IC50:183μM)8)。
(2)代謝酵素阻害
in vitroで、臨床的に意味のある濃度でCYP1A2、CYP2B6、CYP2C8、CYP2C9、CYP2C19、CYP2D6、CYP3A4の活性を阻害しなかった10)。
16.7.2 併用薬がマラビロクの薬物動態に及ぼす影響
(1)CYP3A4又は、CYP3A4及びP-gpを阻害する薬剤のケトコナゾール、リトナビル、サキナビル、ロピナビル・リトナビル、アタザナビル、ダルナビル、テラプレビルは、いずれもマラビロクのCmax及びAUCを増大させた(表2)。CYP3A4誘導薬剤のエファビレンツ、エトラビリン及びリファンピシンはマラビロクのCmax及びAUCを低下させた。[10.2参照]
(2)tipranavir+リトナビル(CYP3A4阻害及びP-gp誘導作用を有する)は、マラビロクの定常状態の薬物動態に影響を及ぼさなかった。
マラビロクの腎クリアランスはCYP3A4阻害剤の非併用時では、総クリアランスの約23%であった7)。腎で消失する薬剤とマラビロクの消失が競合する可能性があるが、トリメトプリム・スルファメトキサゾール合剤(トリメトプリムは腎カチオン輸送を阻害)及びテノホビル(腎で消失)は、マラビロクの薬物動態に影響を及ぼさなかった15)(外国人データ)。
マラビロクの腎クリアランスはCYP3A4阻害剤の非併用時では、総クリアランスの約23%であった7)。腎で消失する薬剤とマラビロクの消失が競合する可能性があるが、トリメトプリム・スルファメトキサゾール合剤(トリメトプリムは腎カチオン輸送を阻害)及びテノホビル(腎で消失)は、マラビロクの薬物動態に影響を及ぼさなかった15)(外国人データ)。
| 併用薬及び用量 | 例数 | マラビロクの用量注) | マラビロクの薬物動態パラメータの比(併用薬の併用時/非併用時)及び90%信頼区間(影響なし=1.00) | ||
| Cmax | AUCtau | Cmin | |||
| CYP3A4又は、CYP3A4及びP-gpを阻害する薬剤 | |||||
| ケトコナゾール 400mg QD16) | 12 | 100mg BID | 3.38 (2.38,4.78) | 5.00 (3.98,6.29) | 3.75 (3.01,4.69) |
| リトナビル 100mg BID16) | 8 | 100mg BID | 1.28 (0.79,2.09) | 2.61 (1.92,3.56) | 4.55 (3.37,6.13) |
| サキナビル(ソフトゲルカプセル)+リトナビル 1000mg+100mg BID16) | 11 | 100mg BID | 4.78 (3.41,6.71) | 9.77 (7.87,12.14) | 11.3 (8.96,14.1) |
| ロピナビル・リトナビル 400mg・100mg BID16) | 11 | 300mg BID | 1.97 (1.66,2.34) | 3.95 (3.43,4.56) | 9.24 (7.98,10.7) |
| アタザナビル 400mg QD16) | 12 | 300mg BID | 2.09 (1.72,2.55) | 3.57 (3.30,3.87) | 4.19 (3.65,4.80) |
| アタザナビル+リトナビル 300mg+100mg QD16) | 12 | 300mg BID | 2.67 (2.32,3.08) | 4.88 (4.40,5.41) | 6.67 (5.78,7.70) |
| ダルナビル+リトナビル 600mg+100mg BID16) | 12 | 150mg BID | 2.29 (1.46,3.59) | 4.05 (2.94,5.59) | 8.00 (6.35,10.1) |
| テラプレビル 750mg TID | 14 | 150mg BID | 7.81 (5.92,10.32) | 9.49 (7.94,11.34) | 10.17 (8.73,11.85) |
| CYP3A4又は、CYP3A4及びP-gpを誘導する薬剤 | |||||
| エファビレンツ 600mg QD16) | 12 | 100mg BID | 0.486 (0.377,0.626) | 0.552 (0.492,0.620) | 0.55 (0.43,0.72) |
| エトラビリン 200mg BID16) | 14 | 300mg BID | 0.400 (0.282,0.566) | 0.468 (0.381,0.576) | 0.609 (0.525,0.707) |
| リファンピシン 600mg QD16) | 12 | 100mg BID | 0.335 (0.260,0.431) | 0.368 (0.328,0.413) | 0.22 (0.17,0.28) |
| ネビラピン注1)(+ラミブジン+テノホビル) 200mg BID(+150mg BID+300mg QD)16) | 8 | 300mg 単回 | 1.54 (0.94,2.51) | 1.01 (0.65,1.55) | − |
| CYP3A4又は、CYP3A4及びP-gpを阻害及び誘導する薬剤 | |||||
| ロピナビル・リトナビル+エファビレンツ 400mg・100mg BID+600mg QD16) | 11 | 300mg BID | 1.25 (1.01,1.55) | 2.53 (2.24,2.87) | 6.29 (4.72,8.39) |
| サキナビル(ソフトゲルカプセル)+リトナビル+エファビレンツ 1000mg+100mg BID+600mg QD16) | 11 | 100mg BID | 2.26 (1.64,3.11) | 5.00 (4.26,5.87) | 8.42 (6.46,10.97) |
| ダルナビル+リトナビル+エトラビリン 600mg+100mg BID+200mg BID16) | 10 | 150mg BID | 1.77 (1.20,2.60) | 3.10 (2.57,3.74) | 5.27 (4.51,6.15) |
| tipranavir+リトナビル 500mg+200mg BID16) | 12 | 150mg BID | 0.86 (0.61,1.21) | 1.02 (0.850,1.23) | 1.80 (1.55,2.09) |
| CYP3A4又は、CYP3A4及びP-gpを阻害及び誘導しない薬剤 | |||||
| ラルテグラビル 400mg BID | 17 | 300mg BID | 0.79 (0.67,0.94) | 0.86 (0.80,0.92) | 0.90 (0.85,0.96) |
16.7.3 マラビロクが併用薬の薬物動態に及ぼす影響
マラビロクはジゴキシン(P糖蛋白の基質)の薬物動態に臨床的に意味のある影響を及ぼさなかった。
マラビロクは、ジドブジン(CYP以外による代謝及び腎で消失)又はラミブジン(主に腎で消失)の薬物動態に影響を及ぼさなかった17)。マラビロクは、ミダゾラム、経口避妊薬(エチニルエストラジオール及びレボノルゲストレル)の薬物動態には臨床的に意味のある影響を及ぼさなかった17)。また、尿中6β-ヒドロキシコルチゾール/コルチゾール比にも影響はなく、マラビロクはin vivoにおいてCYP3A4を誘導しないことが示唆された4)。マラビロクの曝露量が増加した場合にマラビロクがCYP2D6を阻害する可能性は否定できないが、in vitro試験及び臨床試験成績から併用薬の薬物動態に影響を与える可能性は低いものと考えられる(外国人データ)。
マラビロクは、ジドブジン(CYP以外による代謝及び腎で消失)又はラミブジン(主に腎で消失)の薬物動態に影響を及ぼさなかった17)。マラビロクは、ミダゾラム、経口避妊薬(エチニルエストラジオール及びレボノルゲストレル)の薬物動態には臨床的に意味のある影響を及ぼさなかった17)。また、尿中6β-ヒドロキシコルチゾール/コルチゾール比にも影響はなく、マラビロクはin vivoにおいてCYP3A4を誘導しないことが示唆された4)。マラビロクの曝露量が増加した場合にマラビロクがCYP2D6を阻害する可能性は否定できないが、in vitro試験及び臨床試験成績から併用薬の薬物動態に影響を与える可能性は低いものと考えられる(外国人データ)。
注)本剤の承認された用法及び用量は、「通常、成人にはマラビロクとして1回300mgを1日2回経口投与する。なお、投与に際しては必ず他の抗HIV薬を併用し、併用薬に応じて適宜増減すること。」である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 海外第III相臨床試験(A4001027、A4001028)
他の抗HIV薬による治療歴のあるCCR5指向性HIV-1感染患者1076例を対象に、マラビロク(300mg、1日1回又は1日2回)注)又はプラセボと最適背景療法を併用した多施設共同二重盲検試験2試験を実施した結果、以下の成績が得られた18)(外国人データ)。
| マラビロク+OBT注1) 300mg、1日2回 (n=426) | プラセボ+OBT注1) (n=209) | |
| HIV-1 RNA量ベースラインからの変化量(log10 copies/mL) | −1.84 | −0.78 |
| −1.05(−1.33,−0.78)注2) | ||
| HIV-1 RNA量が<400copies/mLとなった症例数の割合 | 56.1% | 22.5% |
| オッズ比:4.76(3.24,7.00)注2) | ||
| HIV-1 RNA量が<50copies/mLとなった症例数の割合 | 45.5% | 16.7% |
| オッズ比:4.49(2.96,6.83)注2) | ||
| CD4陽性リンパ球数ベースラインからの変化量(/mm3) | 124.07 | 60.93 |
| 63.13(44.28,81.99)注2) |
| マラビロク+OBT注1) 300mg、1日2回 (n=426) | プラセボ+OBT注1) (n=209) | ||
| ベースラインHIV-1 RNA量 | <5.0 log10 copies/mL | 58.4% | 26.0% |
| ≧5.0 log10 copies/mL | 34.7% | 9.5% | |
| ベースラインCD4陽性リンパ球数(/mm3) | <50 | 16.5% | 2.6% |
| 50-100 | 36.4% | 12.0% | |
| 101-200 | 56.7% | 21.8% | |
| 201-350 | 57.8% | 21.0% | |
| ≧350 | 72.9% | 38.5% | |
| 併用した抗HIV薬の数注2),注3) | 0 | 32.7% | 2.0% |
| 1 | 44.5% | 7.4% | |
| 2 | 58.2% | 31.7% | |
| ≧3 | 62.0% | 38.6% | |
マラビロク(300mg、1日2回)及び最適背景療法を併用した際にみられた副作用発現頻度は50.0%(213/426例)であった。主な副作用は、悪心12.0%(37/426例)、下痢8.7%(51/426例)、疲労7.3%(31/426例)及び頭痛7.0%(30/426例)であった18)(外国人データ)。
17.1.2 海外臨床試験(A4001029)
他の抗HIV薬による治療歴のあるCXCR4指向性HIV-1感染患者、CCR5/CXCR4二重又は混合指向性HIV-1感染患者を対象に、マラビロク(300mg、1日1回又は1日2回)注)又はプラセボと最適背景療法を併用した多施設共同二重盲検試験を実施した。
その結果、マラビロクはCXCR4指向性、CCR5/CXCR4二重又は混合指向性のHIV-1感染患者において、HIV-1 RNA量及びCD4陽性リンパ球数に対し、有意な影響を及ぼさないことが確認された19)(外国人データ)。
その結果、マラビロクはCXCR4指向性、CCR5/CXCR4二重又は混合指向性のHIV-1感染患者において、HIV-1 RNA量及びCD4陽性リンパ球数に対し、有意な影響を及ぼさないことが確認された19)(外国人データ)。
17.1.3 海外臨床試験(A4001026)
他の抗HIV薬による治療歴のないCCR5指向性HIV-1感染患者を対象に、マラビロク(300mg、1日1回又は1日2回)注)又はエファビレンツと併用薬(ジドブジン300mg及びラミブジン150mg、各1日2回)を投与した多施設共同二重盲検試験を実施した。高精度指向性検査を用いてCCR5指向性HIV-1感染例を選択した結果、以下の成績が得られた(外国人データ)。
| 項目 | マラビロク投与群 (N=311) | エファビレンツ投与群 (N=303) | 両投与群間の差 | ||
| 割合の差 | 片側97.5%CIの下限 | ||||
| 48週後 | |||||
| <400copies/mL | 73.3%(228) | 72.3%(219) | 0.6 | −6.4 | |
| <50copies/mL | 68.5%(213) | 68.3%(207) | −0.2 | −7.4 | |
| 96週後 | |||||
| <400copies/mL | 64.0%(199) | 64.4%(195) | −0.4 | −7.9 | |
| <50copies/mL | 58.8%(183) | 62.7%(190) | −3.9 | −11.5 | |
| 項目 | 48週 | 96週 | ||
| マラビロク投与群 | エファビレンツ投与群 | マラビロク投与群 | エファビレンツ投与群 | |
| レスポンダー | 213例 | 216例 | 188例 | 184例 |
| ノンレスポンダー | 98例 | 87例 | 123例 | 119例 |
| ウイルス学的な治療失敗 | 26% | 9% | 22% | 7% |
| リバウンド | 22% | 14% | 31% | 24% |
| (治療中止例) | ||||
| 有害事象 | 13% | 49% | 15% | 40% |
| 不参加 | 20% | 16% | 20% | 15% |
| 死亡症例 | 1% | 0 | 2% | 2% |
| その他 | 11% | 9% | 11% | 13% |
| 治療 | 治療失敗例での耐性 | Tropism Status | ||
| R5 | X4/DM | NA | ||
| マラビロク投与群 300mg BID (N=39) | チミジン誘導体関連変異 | 5.1% | 5.1% | 0 |
| M184VI注1) | 25.6% | 20.5% | 5.1% | |
| エファビレンツ耐性 | 0 | 0 | 0 | |
| エファビレンツ投与群 600mg QD (N=18) | チミジン誘導体関連変異 | 11.1% | 0 | 0 |
| M184VI注1) | 27.8% | 0 | 16.7% | |
| エファビレンツ耐性 | 38.9% | 0 | 22.2% | |
注)本剤の承認された用法及び用量は、「通常、成人にはマラビロクとして1回300mgを1日2回経口投与する。なお、投与に際しては必ず他の抗HIV薬を併用し、併用薬に応じて適宜増減すること。」である。
18. 薬効薬理
18.1 作用機序
マラビロクは、HIVが細胞に侵入する際に利用する補受容体であるCC Chemokine Receptor 5(CCR5)阻害剤である。マラビロクは、細胞膜上のCCR5に選択的に結合し、HIV-1エンベロープ糖タンパク質gp120とCCR5との相互作用を遮断することにより、CCR5指向性HIV-1の細胞内への侵入を阻害する。なお、マラビロクは、CXCR4指向性及びCCR5/CXCR4二重指向性HIV-1の細胞内への侵入を阻害しない20)。
18.2 抗ウイルス作用
CCR5指向性HIV-1初代臨床分離株43株においてマラビロクの抗ウイルス活性を評価した結果、マラビロクのIC90値はウイルスのサブタイプ間で有意な差はなく、その平均値は血清補正後の非結合型濃度として0.57ng/mLであった。一方、CXCR4使用ウイルス注)に対する抗ウイルス作用は示さなかった。HIV-2に対するマラビロクの抗ウイルス活性は検討されていない20)21)。
ヌクレオシド系逆転写酵素阻害剤(NRTI:アバカビル、ジダノシン、エムトリシタビン、ラミブジン、スタブジン、テノホビル、ザルシタビン、ジドブジン)、非ヌクレオシド系逆転写酵素阻害剤(NNRTI:デラビルジン、エファビレンツ、ネビラピン)、プロテアーゼ阻害剤(アンプレナビル、アタザナビル、インジナビル、ロピナビル、ネルフィナビル、リトナビル、サキナビル)、又はHIV融合阻害剤(enfuvirtide)とマラビロクを併用した場合、抗ウイルス活性に拮抗作用は認められなかった20)。
注)CXCR4使用ウイルス:CXCR4指向性又はCCR5/CXCR4二重指向性ウイルス
18.3 耐性
18.3.1 In vitro試験
CCR5指向性HIV-1臨床分離株2株を連続継代培養した結果、マラビロクに対する感受性が低下した変異株が分離された。これらのマラビロク耐性ウイルスはCCR5指向性を維持しており、CXCR4指向性又はCCR5/CXCR4二重指向性への変化は認められなかった22)。
(1)表現型耐性
マラビロク耐性ウイルスの特徴は、in vitro抗ウイルス作用試験でマラビロクが100%阻害作用を示さないことであった22)。表現型耐性の指標として通常用いられるIC50値は、マラビロクに対する感受性の低下にもかかわらず変動しない場合があり、表現型耐性の判定には有用ではない。
(2)遺伝子型耐性
アミノ酸残基の変異はgp120に集中していた。しかしながら、変異の部位は分離株ごとに異なっており、これらの変異とマラビロク感受性との関連は明らかではない22)。
(3)交差耐性
培養細胞を用いた系で、マラビロクは、NRTI、NNRTI、プロテアーゼ阻害剤及びenfuvirtideに耐性を有するHIV-1臨床分離株に対し、抗ウイルス活性を示した。In vitroで生じたマラビロク耐性ウイルスは、enfuvirtide及びサキナビルに対し、感受性を維持していた23)。
18.3.2 臨床試験
抗HIV薬による治療歴のあるCCR5指向性HIV-1感染患者を対象とした試験(試験A4001027及び試験A4001028)において、スクリーニング期からベースライン時までの間(4〜6週間)で、7.6%の被験者のウイルスの指向性がCCR5指向性からCXCR4指向性又は二重/混合指向性へ変化した23)24)。
また、抗HIV薬による治療歴のない患者を対象とした試験(試験A4001026)において、スクリーニング期からベースライン時までの間(4〜6週間)で、3.6%の被験者のウイルスの指向性がCCR5指向性からCXCR4指向性又は二重/混合指向性へ変化した。
また、抗HIV薬による治療歴のない患者を対象とした試験(試験A4001026)において、スクリーニング期からベースライン時までの間(4〜6週間)で、3.6%の被験者のウイルスの指向性がCCR5指向性からCXCR4指向性又は二重/混合指向性へ変化した。
(1)CXCR4使用ウイルスを伴う治療の失敗
マラビロクによる治療が成功しなかった患者の約60%において、治療失敗時にCXCR4使用ウイルスが検出された。これに対し、プラセボ群(最適背景療法の併用)の治療失敗例でCXCR4使用ウイルスが検出された患者数は6%であった。これらのCXCR4使用ウイルスの起源を検討するため、治療失敗時にCXCR4使用ウイルスが検出された20例(マラビロク群16例、プラセボ群4例)のウイルスのクローン分析を行った結果、CXCR4使用ウイルスは、指向性変異(CCR5指向性ウイルスがCXCR4指向性に変化した)によるのではなく、治療前の指向性検査では検出することのできなかったわずかな量のCXCR4使用ウイルスに由来することが示唆された。
ベースライン時にはCCR5指向性ウイルスを有したがその後CXCR4使用ウイルスが検出され治療が失敗した患者のうち38例で、投与中止後35日間以上の追跡観察を行った。これら38例のうち、最終観察までにCCR5指向性に戻らなかった症例は、3例のみであった。
CXCR4使用ウイルスが検出された治療失敗時の他の抗HIV薬に対する耐性パターンは、ベースライン時のCCR5指向性ウイルスと変わらなかった。したがって、抗HIV薬療法を選択する際には、ベースライン時には検出できないCXCR4使用ウイルスが、ベースライン時に検出されるCCR5指向性ウイルスと同じ耐性パターンを有している可能性を考慮する必要がある23)24)。
ベースライン時にはCCR5指向性ウイルスを有したがその後CXCR4使用ウイルスが検出され治療が失敗した患者のうち38例で、投与中止後35日間以上の追跡観察を行った。これら38例のうち、最終観察までにCCR5指向性に戻らなかった症例は、3例のみであった。
CXCR4使用ウイルスが検出された治療失敗時の他の抗HIV薬に対する耐性パターンは、ベースライン時のCCR5指向性ウイルスと変わらなかった。したがって、抗HIV薬療法を選択する際には、ベースライン時には検出できないCXCR4使用ウイルスが、ベースライン時に検出されるCCR5指向性ウイルスと同じ耐性パターンを有している可能性を考慮する必要がある23)24)。
(2)CCR5指向性ウイルスを伴う治療の失敗
19. 有効成分に関する理化学的知見
19.1. マラビロク
| 一般的名称 | マラビロク |
|---|---|
| 一般的名称(欧名) | Maraviroc |
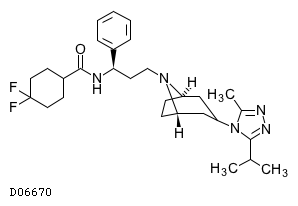
| 化学名 | 4,4-Difluoro-N-[(1S)-3-{(1R,3s,5S)-3-[3-methyl-5-(propan-2-yl)-4H-1,2,4-triazol-4-yl]-8-azabicyclo[3.2.1]octan-8-yl}-1-phenylpropyl]cyclohexanecarboxamide |
| 分子式 | C29H41F2N5O |
| 分子量 | 513.67 |
| 物理化学的性状 | 白色〜微黄色の結晶性の粉末である。メタノールに極めて溶けやすく、アセトニトリル、N,N-ジメチルアセトアミド又はエタノール(99.5)に溶けやすく、水に極めて溶けにくい。 |
| 分配係数 | (log):2.1(pH7.4、1-オクタノール/水系) |
| KEGG DRUG |
|
22. 包装
60錠[瓶、バラ]
23. 主要文献
- 社内資料:安全性薬理試験(2008年12月25日承認、CTD2.6.2.4)
- 社内資料:反復投与毒性試験(2008年12月25日承認、CTD2.6.6.3)
- 社内資料:健康成人を対象とした単回経口投与試験(2008年12月25日承認、CTD2.7.2.2.2)
- Abel S,et al., Br J Clin Pharmacol., 65 (Suppl.1), 5, (2008) »PubMed »DOI
- 社内資料:健康成人及びHIV感染患者の定常状態の薬物動態パラメータ(2008年12月25日承認、CTD2.7.2.2)
- 社内資料:健康成人及びHIV感染患者を対象とした食事の影響の検討(2008年12月25日承認、CTD2.7.1.2.3)
- Abel S,et al., Br J Clin Pharmacol., 65 (Suppl.1), 60, (2008) »PubMed »DOI
- Walker DK,et al., Drug Metab Dispos., 33 (4), 587, (2005) »PubMed »DOI
- 社内資料:蛋白結合に関する検討(2008年12月25日承認、CTD2.6.4.4.3)
- Hyland R,et al., Br J Clin Pharmacol., 66 (4), 498, (2008) »PubMed »DOI
- 社内資料:健康成人及び肝障害患者を対象とした薬物動態試験
- 社内資料:母集団薬物動態の検討(2008年12月25日承認、CTD2.7.2.1.3)
- 社内資料:薬物動態に及ぼす年齢の影響(2008年12月25日承認、CTD2.7.2.3.4.1)
- 社内資料:アジア人及び白人健康成人を対象とした薬物動態試験(2008年12月25日承認、CTD2.7.2.2.4)
- Abel S,et al., Br J Clin Pharmacol., 65 (Suppl.1), 47, (2008) »PubMed »DOI
- 社内資料:薬物相互作用の検討(2008年12月25日承認、CTD2.7.2.2.6)
- Abel S,et al., Br J Clin Pharmacol., 65 (Suppl.1), 19, (2008) »PubMed
- 社内資料:他の抗HIV薬による治療歴のあるCCR5指向性HIV-1感染患者を対象とした多施設共同二重盲検試験
- 社内資料:他の抗HIV薬による治療歴のあるCXCR4指向性HIV-1感染患者及びCCR5/CXCR4二重又は混合指向性HIV-1感染患者を対象とした多施設共同二重盲検試験
- Dorr P,et al., Antimicrob Agents Chemother., 49 (11), 4721, (2005) »PubMed »DOI
- 社内資料:HIV-1初代臨床分離株に対する抗ウイルス活性(2008年12月25日承認、CTD2.6.2.2.1)
- Westby M,et al., J Virol., 81 (5), 2359, (2007) »PubMed »DOI
- 社内資料:耐性及び指向性変化のメカニズムの検討(2008年12月25日承認、CTD2.6.2.2)
- 社内資料:治療失敗例における指向性の検討
- 社内資料:治療失敗例における感受性の検討
- 社内資料:治療失敗例におけるCCR5指向性ウイルスの検討
24. 文献請求先及び問い合わせ先
文献請求先
グラクソ・スミスクライン株式会社
ヴィーブヘルスケア・カスタマー・サービス
東京都港区赤坂1-8-1
電話:0120-066-525(9:00〜17:45/土日祝日及び当社休業日を除く)
製品情報問い合わせ先
グラクソ・スミスクライン株式会社
ヴィーブヘルスケア・カスタマー・サービス
東京都港区赤坂1-8-1
電話:0120-066-525(9:00〜17:45/土日祝日及び当社休業日を除く)
26. 製造販売業者等
26.1 製造販売元
ヴィーブヘルスケア株式会社
東京都港区赤坂1-8-1
26.2 販売元
グラクソ・スミスクライン株式会社
東京都港区赤坂1-8-1