医薬品情報
| 総称名 | サインバルタ |
|---|---|
| 一般名 | デュロキセチン塩酸塩 |
| 欧文一般名 | Duloxetine Hydrochloride |
| 製剤名 | デュロキセチン塩酸塩カプセル |
| 薬効分類名 | セロトニン・ノルアドレナリン再取り込み阻害剤 |
| 薬効分類番号 | 1179 1190 |
| ATCコード | N06AX21 |
| KEGG DRUG |
D01179
塩酸デュロキセチン
|
| JAPIC | 添付文書(PDF) |
添付文書情報2026年5月 改訂(第5版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| サインバルタカプセル20mg | Cymbalta Capsules | 塩野義製薬 | 1179052M1022 | 62.3円/カプセル | 劇薬, 処方箋医薬品 |
| サインバルタカプセル30mg | Cymbalta Capsules | 塩野義製薬 | 1179052M2029 | 77.1円/カプセル | 劇薬, 処方箋医薬品 |
2. 禁忌
次の患者には投与しないこと
4. 効能または効果
5. 効能または効果に関連する注意
<効能共通>
<うつ病・うつ状態>
5.2 本剤を18歳未満の大うつ病性障害患者に投与する際には適応を慎重に検討すること。[9.7参照]
<疼痛の効能共通>
5.3 疼痛に対して本剤を投与する場合は、自殺念慮、自殺企図、敵意、攻撃性等の精神症状の発現リスクを考慮し、本剤の投与の適否を慎重に判断すること。
<線維筋痛症に伴う疼痛>
5.4 線維筋痛症の診断は、米国リウマチ学会の分類(診断)基準等の国際的な基準に基づき慎重に実施し、確定診断された場合にのみ投与すること。
<慢性腰痛症に伴う疼痛>
5.5 最新の診断基準を参考に慢性腰痛症と診断された患者にのみ、本剤の投与を考慮すること。
<変形性関節症に伴う疼痛>
5.6 3ヵ月以上疼痛を有し、最新の診断基準を参考に変形性関節症と診断された患者にのみ、本剤の投与を考慮すること。
6. 用法及び用量
<うつ病・うつ状態、糖尿病性神経障害に伴う疼痛>
通常、成人には1日1回朝食後、デュロキセチンとして40mgを経口投与する。投与は1日20mgより開始し、1週間以上の間隔を空けて1日用量として20mgずつ増量する。
なお、効果不十分な場合には、1日60mgまで増量することができる。
なお、効果不十分な場合には、1日60mgまで増量することができる。
<線維筋痛症に伴う疼痛、慢性腰痛症に伴う疼痛、変形性関節症に伴う疼痛>
通常、成人には1日1回朝食後、デュロキセチンとして60mgを経口投与する。投与は1日20mgより開始し、1週間以上の間隔を空けて1日用量として20mgずつ増量する。
7. 用法及び用量に関連する注意
<うつ病・うつ状態、糖尿病性神経障害に伴う疼痛>
本剤の投与量は必要最小限となるよう、患者ごとに慎重に観察しながら調節すること。
8. 重要な基本的注意
<効能共通>
8.1 うつ症状を呈する患者は希死念慮があり、自殺企図のおそれがあるので、このような患者は投与開始早期並びに投与量を変更する際には患者の状態及び病態の変化を注意深く観察すること。なお、うつ病・うつ状態以外で本剤の適応となる疾患においても自殺企図のおそれがあり、さらにうつ病・うつ状態を伴う場合もあるので、このような患者にも注意深く観察しながら投与すること。[5.1、8.2、8.3、8.4、9.1.5、9.1.6、15.1.1参照]
8.2 不安、焦燥、興奮、パニック発作、不眠、易刺激性、敵意、攻撃性、衝動性、アカシジア/精神運動不穏、軽躁、躁病等があらわれることが報告されている。また、因果関係は明らかではないが、これらの症状・行動を来した症例において、基礎疾患の精神症状の悪化又は自殺念慮、自殺企図、他害行為が報告されている。患者の状態及び病態の変化を注意深く観察するとともに、これらの症状の増悪が観察された場合には、服薬量を増量せず、徐々に減量し、中止するなど適切な処置を行うこと。[5.1、8.1、8.3、8.4、9.1.5、9.1.6、9.1.7、9.1.8、15.1.1参照]
8.3 自殺目的での過量服用を防ぐため、自殺傾向が認められる患者に処方する場合には、1回分の処方日数を最小限にとどめること。[5.1、8.1、8.2、8.4、9.1.5、9.1.6、15.1.1参照]
8.4 家族等に自殺念慮や自殺企図、興奮、攻撃性、易刺激性等の行動の変化及び基礎疾患の精神症状の悪化があらわれるリスク等について十分説明を行い、医師と緊密に連絡を取り合うように指導すること。[5.1、8.1、8.2、8.3、9.1.5、9.1.6、9.1.7、9.1.8、15.1.1参照]
8.5 肝機能障害があらわれることがあるので、適宜肝機能検査(AST、ALT、γ-GTP及び総ビリルビン等)を行うとともに、患者の症状を十分に観察すること。[9.3.2、11.1.5、16.6.2参照]
8.7 眠気、めまい等が起こることがあるので、自動車の運転等危険を伴う機械を操作する際には十分注意させること。また、患者に、これらの症状を自覚した場合は自動車の運転等危険を伴う機械の操作に従事しないよう、指導すること。
8.8 投与中止(特に突然の中止)により、不安、焦燥、興奮、浮動性めまい、錯感覚(電気ショック様感覚を含む)、頭痛、悪心及び筋痛等があらわれることが報告されている。投与を中止する場合には、突然の中止を避け、患者の状態を観察しながら徐々に減量すること。
<糖尿病性神経障害に伴う疼痛>
8.9 本剤による治療は原因療法ではなく対症療法であることから、糖尿病の治療を併せて行うこと。
8.10 本剤の投与により血糖値上昇・HbA1c上昇等、糖尿病が悪化することがあるので、血糖値の推移等を慎重に観察するとともに、必要に応じて糖尿病治療薬の用量調節を行うこと。
<慢性腰痛症に伴う疼痛、変形性関節症に伴う疼痛>
8.11 本剤による治療は原因療法ではなく対症療法であることから、疼痛の原因があればその治療を併せて行い、薬物療法以外の療法も考慮すること。また、患者の状態を十分に観察し、本剤を漫然と投与しないこと。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 前立腺肥大症等排尿困難のある患者
ノルアドレナリン再取り込み阻害作用により症状が悪化することがある。
9.1.2 高血圧又は心疾患のある患者
9.1.3 緑内障又は眼内圧亢進のある患者
症状が悪化することがある。
9.1.4 過度のアルコール摂取者
肝障害が悪化する可能性がある。[10.2参照]
9.1.5 自殺念慮又は自殺企図の既往のある患者、自殺念慮のある患者
9.1.6 躁うつ病患者
9.1.7 脳の器質的障害又は統合失調症の素因のある患者
9.1.8 衝動性が高い併存障害を有する患者
9.1.9 てんかん等の痙攣性疾患又はこれらの既往歴のある患者
痙攣を起こすことがある。
9.1.10 出血性疾患の既往歴又は出血性素因のある患者
出血傾向が増強することがある。[10.2参照]
9.2 腎機能障害患者
9.2.1 高度の腎機能障害のある患者
9.2.2 軽度から中等度の腎機能障害のある患者
本剤の血中濃度が上昇することがある。
9.3 肝機能障害患者
9.3.1 高度の肝機能障害のある患者
投与しないこと。肝機能障害が悪化することがある。また、消失半減期が延長し、本剤の血中濃度が上昇することがある。[2.3参照]
9.3.2 軽度から中等度の肝機能障害のある患者
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断された場合にのみ投与すること。妊娠末期にSNRI、SSRIを投与された女性が出産した新生児において、入院期間の延長、呼吸補助、経管栄養を必要とする、離脱症状と同様の症状が出産直後にあらわれたとの報告がある。臨床所見としては、呼吸窮迫、チアノーゼ、無呼吸、発作、体温調節障害、哺乳障害、嘔吐、低血糖症、筋緊張低下、筋緊張亢進、反射亢進、振戦、ぴくつき、易刺激性、持続性の泣きが報告されている。
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。ラット及びヒトで乳汁中へ移行することが報告されている。[16.3.1参照]
9.7 小児等
海外で実施された7〜17歳の大うつ病性障害(DSM-IV-TR※における分類)患者を対象としたプラセボ対照の臨床試験において有効性が確認できなかったとの報告がある。[5.2参照]
※:DSM-IV-TR:American Psychiatric Association(米国精神医学会)のDiagnostic and Statistical Manual of Mental Disorders.4th edition,Text Revision(DSM-IV-TR精神疾患の診断・統計マニュアル)
9.8 高齢者
10. 相互作用
相互作用序文
本剤の代謝には主として肝代謝酵素CYP1A2が関与し、CYP2D6も一部寄与している。また、本剤はCYP2D6を競合的に阻害する。
薬物代謝酵素用語
CYP1A2
薬物代謝酵素用語
CYP2D6
10.1 併用禁忌
| モノアミン酸化酵素(MAO)阻害剤 セレギリン塩酸塩 (エフピー) ラサギリンメシル酸塩 (アジレクト) サフィナミドメシル酸塩 (エクフィナ) [2.2参照] | 他の抗うつ剤で併用により発汗、不穏、全身痙攣、異常高熱、昏睡等の症状があらわれたとの報告がある。 MAO阻害剤の投与を受けた患者に本剤を投与する場合には、少なくとも2週間の間隔をおき、また、本剤からMAO阻害剤に切り替えるときは5日間の間隔をおくこと。 | 主にMAO阻害剤による神経外アミン総量の増加及び抗うつ剤によるモノアミン作動性神経終末におけるアミン再取り込み阻害によると考えられる。 |
10.2 併用注意
| ピモジド | QT延長、心室性不整脈(Torsade de pointesを含む)等の心血管系副作用が発現することがあるので注意すること。 | 本剤は、ピモジドの肝での酸化的代謝を阻害し、血中濃度を上昇させると考えられる。 |
| アルコール [9.1.4参照] | 相互に中枢神経抑制作用を増強することがあるので注意すること。また、肝機能が悪化するおそれがある。 | アルコールは中枢神経抑制作用を有する。また、過度のアルコール摂取と本剤との併用により、肝機能が悪化することがある。 |
| 中枢神経抑制剤 バルビツール酸誘導体、ロラゼパム等 | 相互に作用を増強することがあるので、本剤及びこれらの薬剤の用量を減量するなど注意して投与すること。 | 機序は不明 |
| メチルチオニニウム塩化物水和物(メチレンブルー) | セロトニン症候群があらわれるおそれがある。 | 左記薬剤のMAO阻害作用によりセロトニン作用が増強される。 |
| フルボキサミンマレイン酸塩、シプロフロキサシン、エノキサシン等 [16.7.1参照] | 本剤の血中濃度が上昇することがあるので、本剤の用量を減量するなど注意して投与すること。 | これらの薬剤のCYP1A2阻害作用により、本剤の血中濃度が上昇することがある。 本剤とフルボキサミンとの併用により、本剤の血漿クリアランスが減少したとの報告がある。 |
| 三環系抗うつ剤 アミトリプチリン塩酸塩、ノルトリプチリン塩酸塩、イミプラミン塩酸塩等 フェノチアジン系抗精神病剤 ペルフェナジン 抗不整脈剤 プロパフェノン塩酸塩、フレカイニド酢酸塩 | これらの薬剤の血中濃度が上昇することがあるので、これらの薬剤の用量を減量するなど注意して投与すること。 | 本剤のCYP2D6阻害作用により、これらの薬剤の血中濃度が上昇することがある。 本剤とCYP2D6基質であるデシプラミンとの併用により、デシプラミンのAUCが増加したとの報告がある。 |
| パロキセチン塩酸塩水和物、キニジン硫酸塩水和物等 [16.7.1参照] | 本剤の血中濃度が上昇することがあるので、本剤の用量を減量するなど注意して投与すること。 | これらの薬剤のCYP2D6阻害作用により、本剤の血中濃度が上昇することがある。 本剤とパロキセチンとの併用により、本剤の血漿クリアランスが減少したとの報告がある。 |
| セロトニン作用薬 炭酸リチウム、セロトニン・ノルアドレナリン再取り込み阻害剤(SNRI)及び選択的セロトニン再取り込み阻害剤(SSRI)、トラマドール塩酸塩、トリプタン系薬剤、L-トリプトファン含有製剤、リネゾリド等 セイヨウオトギリソウ(セント・ジョーンズ・ワート)含有食品等 [11.1.1参照] | 相互にセロトニン作用を増強することによりセロトニン症候群等のセロトニン作用による症状があらわれることがあるので、本剤及びこれらの薬剤の用量を減量するなど注意して投与すること。 | 本剤はセロトニン再取り込み阻害作用を有するため、併用により、セロトニン作用が増強することがある。 |
| 降圧剤 クロニジン塩酸塩等 | 降圧剤の作用を減弱することがあるので、本剤の用量を減量もしくはこれらの薬剤を増量するなど注意して投与すること。 | 本剤のノルアドレナリン再取り込み阻害作用によると考えられる。 |
| アドレナリン、ノルアドレナリン | これらの薬剤(特に注射剤)との併用により、心血管作用(血圧上昇等)が増強することがあるので、本剤及びこれらの薬剤の用量を減量するなど注意して投与すること。 | 本剤はノルアドレナリン再取り込み阻害作用を有するため、併用により、アドレナリン作用が増強することがある。 |
| 血漿蛋白との結合率の高い薬剤 ワルファリンカリウム等 | 相互に作用を増強することがあるので、本剤及びこれらの薬剤の用量を減量するなど注意して投与すること。 | 本剤は血漿蛋白との結合率が高いため、併用により、本剤及びこれらの薬剤の血中遊離濃度が上昇することがある。 |
| 出血傾向が増強する薬剤 非定型抗精神病剤、フェノチアジン系薬剤、三環系抗うつ剤、アスピリン等の非ステロイド系抗炎症剤、ワルファリンカリウム等 [9.1.10参照] | 出血傾向が増強することがあるので、本剤及びこれらの薬剤の用量を減量するなど注意して投与すること。 | SNRI、SSRIとこれらの薬剤との併用により、出血傾向が増強すると考えられる。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には、必要に応じて、減量、休薬又は中止するなどの適切な処置を行うこと。
11.1.1 セロトニン症候群(頻度不明)
不安、焦燥、興奮、錯乱、発汗、下痢、発熱、高血圧、固縮、頻脈、ミオクローヌス、自律神経不安定等があらわれることがある。セロトニン作用薬との併用時に発現する可能性が高くなるため、特に注意すること。異常が認められた場合には投与を中止し、体冷却、水分補給等の全身管理と共に適切な処置を行うこと。[10.2参照]
11.1.2 悪性症候群(頻度不明)
発熱、無動緘黙、強度の筋強剛、嚥下困難、頻脈、血圧の変動、発汗、白血球数増加、血清CK(CPK)上昇等の異常が認められた場合には、投与を中止し、体冷却、水分補給等の全身管理と共に適切な処置を行うこと。また、ミオグロビン尿を伴う腎機能の低下がみられ、急性腎障害に至ることがあるので注意すること。
11.1.3 抗利尿ホルモン不適合分泌症候群(SIADH)(頻度不明)
低ナトリウム血症、低浸透圧血症、尿中ナトリウム排泄量の増加、高張尿、痙攣、意識障害等を伴う抗利尿ホルモン不適合分泌症候群(SIADH)があらわれることがあるので、異常が認められた場合には投与を中止し、水分摂取の制限等適切な処置を行うこと。[9.8参照]
11.1.4 痙攣(0.1%未満)、幻覚(頻度不明)
11.1.5 肝機能障害(0.1%未満)、肝炎(頻度不明)、黄疸(頻度不明)
11.1.6 皮膚粘膜眼症候群(Stevens-Johnson症候群)(頻度不明)
11.1.7 アナフィラキシー反応(頻度不明)
呼吸困難、痙攣、血管性浮腫、蕁麻疹等を伴うアナフィラキシー反応があらわれることがある。
11.1.9 尿閉(頻度不明)
症状があらわれた場合には投与を中止し、導尿を実施するなど適切な処置を行うこと。
11.2 その他の副作用
| 5%以上 | 1〜5%未満 | 1%未満 | 頻度不明 | |
| 過敏症注 | 発疹、そう痒、蕁麻疹 | 接触性皮膚炎、光線過敏反応、血管性浮腫、皮膚血管炎 | ||
| 全身症状 | 倦怠感 | ほてり、発熱、悪寒、脱水、脱力感 | ||
| 精神神経系 | 傾眠(24.3%)、頭痛、めまい | 不眠、立ちくらみ、しびれ感、振戦、浮遊感 | あくび、焦燥感、気分高揚、注意力障害、錐体外路症状、不安、異常夢(悪夢を含む)、頭がぼーっとする、性欲減退、躁病反応、錯感覚、無感情、味覚異常 | 激越、オーガズム異常、嗜眠、睡眠障害、歯軋り、失見当識、攻撃性、怒り、歩行障害、開口障害、下肢静止不能症候群、異常感 |
| 消化器 | 悪心(22.4%)、食欲減退、口渇(12.8%)、便秘(12.4%)、下痢 | 腹部痛、嘔吐、腹部膨満感、腹部不快感、消化不良、胃炎 | 口内炎、歯痛、胃腸炎、咽頭不快感 | 咽頭炎、咽喉緊張、口臭、嚥下障害、顕微鏡的大腸炎 |
| 感覚器 | 耳鳴 | 視調節障害、眼乾燥、霧視、耳痛 | 散瞳、緑内障 | |
| 循環器 | 動悸、頻脈、血圧上昇 | 起立性低血圧、上室性不整脈、失神 | ||
| 肝臓 | AST上昇、ALT上昇、γ-GTP上昇、総ビリルビン上昇、Al-P上昇、LDH上昇 | |||
| 血液 | ヘモグロビン減少、赤血球減少、ヘマトクリット減少、鼻出血 | 異常出血(斑状出血、胃腸出血等)、白血球減少 | ||
| 筋・骨格系 | 背部痛、関節痛、筋痛、肩こり、筋痙攣 | 筋緊張 | ||
| 泌尿器・生殖器 | 排尿困難 | 性機能異常(月経異常、射精障害、勃起障害等)、排尿障害、血中クレアチニン上昇、BUN上昇、頻尿、尿中アルブミン/クレアチニン比上昇、尿流量減少 | 多尿、閉経期症状、精巣痛 | |
| 代謝・内分泌 | 高血糖、トリグリセリド上昇、総コレステロール上昇、尿中蛋白陽性 | 血中カリウム減少 | 甲状腺機能低下、低ナトリウム血症、乳汁漏出症、高プロラクチン血症、血中カリウム上昇 | |
| その他 | 発汗、体重減少、体重増加、CK(CPK)上昇 | 浮腫、冷感、熱感、呼吸苦、胸痛、冷汗、咳嗽 |
13. 過量投与
13.1 症状
海外において、本剤3000mgを超える(単剤又は他剤との併用)過量投与が報告されている。過量投与による徴候及び症状は傾眠、昏睡、セロトニン症候群、発作、嘔吐、頻脈であった。
13.2 処置
特異的な解毒剤は知られていない。必要に応じて、活性炭投与等の適切な処置を行なうこと。本剤は分布容積が大きいので、強制利尿、血液潅流、交換輸血はあまり効果的ではない。
14. 適用上の注意
14.1 薬剤交付時の注意
14.1.1 PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
14.1.2 本剤は高温多湿を避けて保存するよう指導すること。
14.1.3 腸溶性コーティングを施しているため、カプセルの内容物を砕いたり、すりつぶしたりしないで服用するよう指導すること。原薬が酸に不安定であり、胃酸で失活することがある。
15. その他の注意
15.1 臨床使用に基づく情報
15.1.1 海外で実施された大うつ病性障害等の精神疾患を有する患者を対象とした、本剤を含む複数の抗うつ剤の短期プラセボ対照臨床試験の検討結果において、24歳以下の患者では、自殺念慮や自殺企図の発現のリスクが抗うつ剤投与群でプラセボ群と比較して高かった。なお、25歳以上の患者における自殺念慮や自殺企図の発現のリスクの上昇は認められず、65歳以上においてはそのリスクが減少した。[5.1、8.1、8.2、8.3、8.4、9.1.5、9.1.6参照]
15.1.2 主に50歳以上を対象に実施された海外の疫学調査において、選択的セロトニン再取り込み阻害剤及び三環系抗うつ剤を含む抗うつ剤を投与された患者で、骨折のリスクが上昇したとの報告がある。
16. 薬物動態
16.1 血中濃度
16.1.1 健康成人
(1)単回投与
健康成人男性(8例)にデュロキセチン10mg、20mg、40mgを食後単回経口投与したときの血漿中濃度及び薬物動態パラメータを図16-1・表16-1に示す。
Cmax及びAUCは用量の増加に従い増大した。Tmax及びT1/2(β)は10〜40mgの用量範囲でほぼ一定であった1)。
Cmax及びAUCは用量の増加に従い増大した。Tmax及びT1/2(β)は10〜40mgの用量範囲でほぼ一定であった1)。
図16-1 食後単回経口投与時の血漿中濃度
| 投与量(mg) | 例数 | Cmax(ng/mL) | Tmax(hr) | AUC0-48(ng・hr/mL) | T1/2(β)(hr) |
| 10 | 8 | 12.08±10.09 | 7.8±2.3 | 155.51±94.64 | 12.75±5.88注 |
| 20 | 18.31±10.89 | 7.5±1.4 | 259.33±141.84 | 15.34±5.87 | |
| 40 | 38.65±19.46 | 6.9±2.0 | 551.75±239.64 | 10.56±2.86 |
(2)反復投与
健康成人男性(各6例)にデュロキセチン20mg2)、40mg3)、60mg4)を1日1回7日間、食後反復経口投与したときの血漿中濃度及び薬物動態パラメータを図16-2・表16-2に示す。
血漿中濃度推移は反復投与により上昇し7日目におけるCmax、AUCは初回投与時と比べて増大したが、投与7日目には定常状態に達していた。
血漿中濃度推移は反復投与により上昇し7日目におけるCmax、AUCは初回投与時と比べて増大したが、投与7日目には定常状態に達していた。
図16-2 食後反復経口投与時の血漿中濃度
| 投与量(mg) | 例数 | Cmax(ng/mL) | Tmax(hr) | AUC0-24(ng・hr/mL) | T1/2(β)(hr) | |
| 20 | 6 | 1日目 | 13.57±4.40 | 6.2±1.0 | 139.56±27.40 | 12.30±3.11 |
| 7日目 | 16.24±4.95 | 6.0±0.0 | 205.32±45.34 | 12.09±2.58 | ||
| 40 | 6 | 1日目 | 22.17±12.67 | 6.7±2.9 | 254.15±151.73 | 13.78±6.82 |
| 7日目 | 31.50±16.81 | 5.8±1.2 | 426.76±263.55 | 17.26±2.25 | ||
| 60 | 6 | 1日目 | 46.2±25.7 | 5.8±1.2 | 519.1±267.4 | 13.46±5.03 |
| 7日目 | 68.1±20.8 | 5.7±0.5 | 895.8±344.3 | 13.18±2.26 |
16.2 吸収
16.2.1 食事の影響
健康成人男性(7例)にデュロキセチン20mgを空腹時あるいは食後に単回経口投与し、食事の影響を検討したときの薬物動態パラメータ及び統計解析結果を表16-3に示す。食後投与のCmaxは空腹時に比べ高い値を示し、有意差が認められたものの、Tmax、AUC、T1/2(β)、Ae(尿中排泄量)は有意な変化を示さなかった5)。
| 投与量(mg) | 例数 | Cmax(ng/mL) | Tmax(hr) | AUC0-48(ng・hr/mL) | T1/2(β)(hr) | Ae0-48(μg) | |
| 20 | 空腹時 | 7 | 8.53±4.12 | 5.7±0.8 | 116.33±58.16 | 9.01±1.42 | 11.36±7.04 |
| 食後 | 10.97±6.17 | 6.0±0.0 | 133.82±66.72 | 9.27±0.79 | 11.93±6.06 | ||
| p値 | 0.0422* | 0.2856 | 0.1427 | 0.7171 | 0.9499 | ||
16.2.2 食事の影響及び投与時間の影響
健康成人女性(12例)を対象に、デュロキセチン40mgを朝空腹時、朝食後、あるいは夜就寝時(空腹)にそれぞれ単回経口投与し、食事の影響及び投与時間の影響を検討したときの薬物動態パラメータ及び統計解析結果を表16-4に示す。
Cmax、AUCは朝食後投与と朝空腹時投与との間で有意差は認められなかった。朝食後投与のTmaxは朝空腹時投与に比べ延長し、有意差が認められた。朝食後投与における血漿中濃度の消失速度定数(λz)は空腹時に比べ大きく、有意差が認められた。
夜就寝時(空腹)投与のCmax、AUCは朝空腹時投与に比べ低く、Tmaxは延長し、それぞれ有意差が認められた6)(外国人によるデータ)。
Cmax、AUCは朝食後投与と朝空腹時投与との間で有意差は認められなかった。朝食後投与のTmaxは朝空腹時投与に比べ延長し、有意差が認められた。朝食後投与における血漿中濃度の消失速度定数(λz)は空腹時に比べ大きく、有意差が認められた。
夜就寝時(空腹)投与のCmax、AUCは朝空腹時投与に比べ低く、Tmaxは延長し、それぞれ有意差が認められた6)(外国人によるデータ)。
| 薬物動態パラメータ | Cmax注2(ng/mL) | Tmax注3(hr) | AUC0-∞注2(ng・hr/mL) | λz(hr−1) | T1/2注4(hr) | |
| 朝空腹時 | 1回目 | 27.5±8.3 | 6.0(4.0-10.0) | 464.3±148.9 | 0.058±0.013 | 11.9(8.2-17.5) |
| 2回目 | 25.9±9.4 | 6.0(1.0-10.0) | 456.7±185.5 | 0.061±0.013 | 11.3(8.0-14.9) | |
| 朝食後 | 24.1±11.4 | 10.0(6.0-16.1) | 402.3±164.5 | 0.070±0.018 | 9.8(5.9-14.1) | |
| 夜就寝時(空腹) | 19.6±6.8 | 10.0(4.0-16.0) | 381.7±154.4 | 0.064±0.011 | 10.8(8.1-16.3) | |
| 朝空腹注1vs.朝食後 p値 | 0.405 | <0.001* | 0.060 | 0.004* | − | |
| 朝空腹注1vs.就寝時 p値 | <0.001* | <0.001* | 0.005* | 0.368 | − | |
16.3 分布
16.3.1 乳汁移行
16.3.2 胎児への移行(参考)
(1)胎児移行
妊娠第12日目のラット(n=3〜4)に14C-標識デュロキセチン塩酸塩(デュロキセチンとして45mg/kg)を経口投与したときの放射能の胎児移行率は投与量の0.02%以下であった7)。
(2)胎児主要組織への移行
妊娠第18日目のラット(n=1)に14C-標識デュロキセチン塩酸塩(デュロキセチンとして45mg/kg)を経口投与したとき、胎児主要組織への放射能の移行が認められたが、投与後24時間では検出限界以下まで低下した7)。
16.3.3 蛋白結合率
健康成人を対象とした単回及び反復投与試験におけるex vivoの血清蛋白結合率を測定した結果、97〜99%であった。結合率は血漿中デュロキセチン濃度に依存せず、反復投与による変化は認められなかった1)。
16.4 代謝
ヒト肝ミクロソームを用いた試験(発現CYPでの代謝と特異的阻害剤による阻害)の結果より、ヒト肝ミクロソームでは主に4位及び5位の水酸化が起こり、その反応にはCYP1A2及びCYP2D6が関与していると考えられる9)(in vitro試験)。
デュロキセチンの主代謝物は、4-ヒドロキシ デュロキセチン グルクロナイドで、他に5-ヒドロキシ 6-メトキシ デュロキセチン サルフェート、5,6-ジヒドロキシ デュロキセチン グルクロナイド、6-ヒドロキシ 5-メトキシ デュロキセチン グルクロナイドが認められ、いずれもデュロキセチンが酸化された後、抱合を受けた代謝物であった6)(外国人によるデータ)。
デュロキセチンの主代謝物は、4-ヒドロキシ デュロキセチン グルクロナイドで、他に5-ヒドロキシ 6-メトキシ デュロキセチン サルフェート、5,6-ジヒドロキシ デュロキセチン グルクロナイド、6-ヒドロキシ 5-メトキシ デュロキセチン グルクロナイドが認められ、いずれもデュロキセチンが酸化された後、抱合を受けた代謝物であった6)(外国人によるデータ)。
16.5 排泄
糞中及び尿中にデュロキセチンはほとんど存在せず、投与量の72.0%は代謝物として尿中に排泄され、18.5%は糞中に排泄された6)(外国人によるデータ)。
16.6 特定の背景を有する患者
16.6.1 腎障害患者
16.6.2 肝障害患者
16.6.3 高齢者
16.7 薬物相互作用
16.7.1 本剤が受ける影響
(1)フルボキサミン
(2)パロキセチン
(3)ファモチジン、活性炭
健康成人男性(14例)に、デュロキセチン(40mg朝空腹時単回経口投与)とファモチジン40mg(朝空腹時単回経口投与)、活性炭(活性炭液剤として50g朝空腹時単回経口投与)をそれぞれ併用投与し、本剤の薬物動態を評価した。本剤の吸収に及ぼすファモチジンの影響は小さかった。活性炭の併用により、本剤のCmax、AUCはそれぞれ68%及び65%に低下し、T1/2は0.91倍に短縮し、いずれも有意差が認められた8)(外国人によるデータ)。
16.7.2 他剤に及ぼす影響
テオフィリン
健康成人男性(10例)に、デュロキセチン(60mg/日2回反復経口投与)とテオフィリン(アミノフィリンとして250mgの30分間点滴静脈内投与)を併用投与し、テオフィリンの薬物動態を評価した。テオフィリン薬物動態に有意な変化はみられなかった8)(外国人によるデータ)。
16.7.3 相互に及ぼす影響
ロラゼパム
健康成人(男性8例、女性8例)に、デュロキセチン(60mg/日2回反復経口投与)とロラゼパム(2mg/日2回反復経口投与)を併用投与し、相互に及ぼす影響を評価した。薬物動態に相互作用はみられなかった8)(外国人によるデータ)。
注:本剤の承認された用法は1日1回朝食後に経口投与、1日最大用量は60mgである。
17. 臨床成績
17.1 有効性及び安全性に関する試験
承認時における臨床試験成績の概要は以下のとおりであった。
<うつ病・うつ状態>
17.1.1 国内第III相 二重盲検並行群間比較試験
うつ病・うつ状態の患者を対象として、本剤(デュロキセチンとして40mg又は60mg)、プラセボ又はパロキセチン塩酸塩水和物(パロキセチンとして20〜40mg)を6週間投与した結果、主要評価指標であるハミルトンうつ病評価尺度(HAM-D17)合計評点の変化量は表17-1のとおりであり、本剤(40mg及び60mg併合群)のプラセボに対する優越性が示された。また、本剤40mg群と60mg群で用量反応関係は認められなかった11)。
| 投与群 | 例数 | HAM-D17合計評点 | 変化量 | ||||
| ベースライン注1 | 最終評価時 | ベースラインからの変化量 | プラセボ群との対比較注2 | ||||
| 群間差(95%信頼区間) | p値 | ||||||
| プラセボ群 | 145 | 20.4±4.2 | 12.2±7.0 | −8.3±5.8 | − | − | |
| 本剤 | 40mg群 | 73 | 20.6±4.4 | 10.1±5.6 | −10.5±5.7 | −2.17(−3.83,−0.52) | 0.0103* |
| 60mg群 | 74 | 20.4±4.1 | 10.5±6.2 | −10.0±6.4 | −1.70(−3.35,−0.05) | 0.0440* | |
| 併合群 | 147 | 20.5±4.2 | 10.3±5.9 | −10.2±6.1 | −1.93(−3.28,−0.58) | 0.0051* | |
| パロキセチン群 | 148 | 20.4±4.8 | 11.0±7.4 | −9.4±6.9 | −1.29(−2.64,0.07) | 0.0623 | |
副作用発現頻度は81.7%(143/175例)であった。主な副作用は悪心26.3%(46/175例)、傾眠21.1%(37/175例)、頭痛17.7%(31/175例)であった11)。
17.1.2 国内第III相 長期投与試験
うつ病・うつ状態の患者を対象として、本剤(デュロキセチンとして40mg又は60mg)を最大52週間投与した結果、HAM-D17合計評点の変化量は表17-2のとおりであり、長期間にわたり抗うつ効果が維持された。また、本剤40mgから60mgへの増量により改善した症例も認められた12)。
| 評価時期 | 例数 | HAM-D17合計評点 | 変化量 |
| ベースライン | 215 | 20.9±5.1 | − |
| 6週時 | 187 | 12.5±5.3 | −8.3±5.2 |
| 12週時 | 182 | 10.1±5.2 | −10.6±5.6 |
| 24週時 | 172 | 8.4±5.3 | −12.6±6.5 |
| 52週時 | 146 | 5.5±4.8 | −15.6±6.1 |
副作用発現頻度は93.0%(200/215例)であった。主な副作用は悪心32.1%(69/215例)、傾眠29.3%(63/215例)、口渇22.3%(48/215例)、頭痛21.9%(47/215例)、下痢15.8%(34/215例)、便秘13.5%(29/215例)、トリグリセライド増加13.0%(28/215例)であった12)。
<糖尿病性神経障害に伴う疼痛>
17.1.3 国内第III相 二重盲検並行群間比較試験
糖尿病性神経障害に伴う疼痛の患者を対象として、本剤(デュロキセチンとして40mg又は60mg)又はプラセボを12週間投与した結果、主要評価指標である24時間平均疼痛重症度スコア週平均値の変化量は表17-3のとおりであり、本剤(40mg及び60mg併合群)のプラセボに対する優越性が示された。また、本剤40mg群と60mg群で用量反応関係は認められなかった13)。
| 投与群 | 24時間平均疼痛重症度スコア週平均値 | 変化量 | ||||
| ベースライン注1 | 投与12週時注1 | ベースラインからの変化量注2 | プラセボ群との対比較 | |||
| 群間差(95%信頼区間) | p値 | |||||
| プラセボ群 | 5.78±1.17(167) | 4.38±1.99(150) | −1.61±0.18 | − | − | |
| 本剤 | 40mg群 | 5.79±1.23(85) | 3.54±1.86(73) | −2.41±0.21 | −0.81(−1.18,−0.43) | − |
| 60mg群 | 5.76±1.17(86) | 3.41±1.77(72) | −2.53±0.21 | −0.93(−1.30,−0.56) | − | |
| 併合群 | 5.77±1.20(171) | 3.48±1.81(145) | −2.47±0.18 | −0.87(−1.17,−0.56) | <0.0001* | |
副作用発現頻度は62.0%(106/171例)であった。主な副作用は傾眠21.6%(37/171例)、悪心14.0%(24/171例)、便秘5.3%(9/171例)、倦怠感5.3%(9/171例)であった13)。
17.1.4 国内第III相 長期投与試験
糖尿病性神経障害に伴う疼痛の患者を対象として、本剤(デュロキセチンとして40mg又は60mg)を最大51週間投与した結果、簡易疼痛調査一覧(BPI)−疼痛重症度(平均の痛み)スコアの変化量は表17-4のとおりであり、長期間にわたり鎮痛効果が維持された14)。
| 評価時期 | 例数 | BPI-疼痛重症度(平均の痛み)スコア | 変化量 |
| ベースライン | 258 | 3.9±1.9 | − |
| 8週時 | 245 | 2.6±1.7 | −1.3±1.4 |
| 16週時 | 230 | 2.4±1.8 | −1.5±1.5 |
| 28週時 | 214 | 2.1±1.7 | −1.8±1.7 |
| 50/51週時注 | 191 | 1.8±1.4 | −2.2±1.6 |
副作用発現頻度は67.1%(173/258例)であった。主な副作用は傾眠11.2%(29/258例)、HbA1c増加9.3%(24/258例)、便秘8.1%(21/258例)、悪心6.6%(17/258例)であった15)。
<線維筋痛症に伴う疼痛>
17.1.5 国内第III相 二重盲検並行群間比較試験
線維筋痛症患者を対象として、本剤(デュロキセチンとして60mg)又はプラセボを14週間投与した結果、主要評価指標であるBPI-疼痛重症度(平均の痛み)スコアの14週時変化量は表17-5のとおりであり、主要解析(混合効果モデルによる解析)において、本剤60mgのプラセボに対する優越性は示されなかった。なお、副次解析であるLOCF(Last Observation Carried Forward)法により14週時の欠測値を補完した共分散分析では、群間に有意差が認められた16)。
| 解析手法 | 投与群 | BPI-疼痛重症度(平均の痛み)スコア | 変化量 | |||
| ベースライン注1 | 投与14週時注1 | ベースラインからの変化量注2 | プラセボ群との対比較 | |||
| 群間差(95%信頼区間) | p値 | |||||
| 主要解析:混合効果モデルによる解析 | プラセボ群 | 6.13±1.35(195) | 4.33±1.97(147) | −1.58±0.23 | − | − |
| 60mg群 | 6.05±1.29(191) | 3.88±1.84(163) | −1.90±0.23 | −0.32(−0.70,0.06) | 0.0988 | |
| 副次解析:共分散分析(LOCF) | プラセボ群 | 6.13±1.35(195) | 4.55±2.02(195) | −1.22±0.26 | − | − |
| 60mg群 | 6.05±1.29(191) | 4.13±1.94(191) | −1.60±0.26 | −0.38(−0.74,−0.02) | 0.0408* | |
副作用発現頻度は64.4%(125/194例)であった。主な副作用は傾眠25.8%(50/194例)、悪心21.6%(42/194例)、便秘13.9%(27/194例)、口渇6.7%(13/194例)、食欲減退6.7%(13/194例)であった17)。
17.1.6 国内第III相 長期投与試験
線維筋痛症患者を対象として、本剤(デュロキセチンとして60mg)を50週間投与した結果、BPI-疼痛重症度(平均の痛み)スコアの変化量は表17-6のとおりであり、長期間にわたり鎮痛効果が維持された18)。
| 評価時期 | 例数 | BPI-疼痛重症度(平均の痛み)スコア | 変化量 |
| ベースライン | 148 | 4.54±1.99 | − |
| 8週時 | 148 | 3.50±2.01 | −1.04±1.56 |
| 16週時 | 146 | 3.45±2.17 | −1.09±1.77 |
| 28週時 | 134 | 3.32±2.10 | −1.19±1.59 |
| 50週時 | 115 | 3.27±2.34 | −1.31±1.70 |
副作用発現頻度は63.8%(95/149例)であった。主な副作用は傾眠21.5%(32/149例)、便秘16.1%(24/149例)、悪心11.4%(17/149例)、体重増加7.4%(11/149例)、口渇6.7%(10/149例)、倦怠感5.4%(8/149例)であった18)。
<慢性腰痛症に伴う疼痛>
17.1.7 国内第III相 二重盲検並行群間比較試験
非ステロイド性抗炎症薬(NSAIDs)の効果が不十分な慢性腰痛症患者を対象として、本剤(デュロキセチンとして60mg)又はプラセボを14週間投与した結果、主要評価指標であるBPI-疼痛重症度(平均の痛み)スコアの14週時変化量は表17-7のとおりであり、本剤60mg群のプラセボに対する優越性が示された19)。
| 投与群 | BPI-疼痛重症度(平均の痛み)スコア | 変化量 | |||
| ベースライン注1 | 投与14週時注1 | ベースラインからの変化量注2 | プラセボ群との対比較 | ||
| 群間差(95%信頼区間) | p値 | ||||
| プラセボ群 | 5.09±1.04(226) | 3.16±1.78(200) | −1.96±0.11 | − | − |
| 60mg群 | 5.14±1.11(230) | 2.73±1.69(209) | −2.43±0.11 | −0.46(−0.77,−0.16) | 0.0026* |
副作用発現頻度は48.3%(113/234例)であった。主な副作用は傾眠18.8%(44/234例)、便秘10.3%(24/234例)、悪心8.5%(20/234例)であった20)。
17.1.8 国内第III相 長期投与試験
慢性腰痛症患者を対象として、本剤(デュロキセチンとして60mg)を最大50週間投与した結果、BPI-疼痛重症度(平均の痛み)スコアの変化量は表17-8のとおりであり、長期間にわたり鎮痛効果が維持された21)。
| 評価時期 | 例数 | BPI-疼痛重症度(平均の痛み)スコア | 変化量 |
| ベースライン | 150 | 3.89±1.55 | − |
| 8週時 | 142 | 2.35±1.72 | −1.56±1.58 |
| 16週時 | 140 | 2.17±1.71 | −1.76±1.78 |
| 28週時 | 137 | 1.95±1.54 | −2.01±1.76 |
| 50週時 | 121 | 1.59±1.50 | −2.26±1.63 |
副作用発現頻度は50.3%(76/151例)であった。主な副作用は傾眠18.5%(28/151例)、悪心10.6%(16/151例)、便秘8.6%(13/151例)、口渇6.0%(9/151例)であった21)。
<変形性関節症に伴う疼痛>
17.1.9 国内第III相 二重盲検並行群間比較試験
試験開始前の3ヵ月間で月に14日以上の痛みを有する特発性変形性膝関節症患者を対象として、本剤(デュロキセチンとして60mg)又はプラセボを14週間投与した結果、主要評価指標であるBPI-疼痛重症度(平均の痛み)スコアの14週時変化量は表17-9のとおりであり、本剤60mg群のプラセボに対する優越性が示された22)。
| 投与群 | BPI-疼痛重症度(平均の痛み)スコア | 変化量 | |||
| ベースライン注1 | 投与14週時注1 | ベースラインからの変化量注2 | プラセボ群との対比較 | ||
| 群間差(95%信頼区間) | p値 | ||||
| プラセボ群 | 5.06±0.98(176) | 3.14±1.70(161) | −1.80±0.12 | − | − |
| 60mg群 | 5.03±0.96(177) | 2.44±1.54(160) | −2.57±0.12 | −0.77(−1.11,−0.43) | <0.0001* |
副作用発現頻度は43.3%(77/178例)であった。主な副作用は傾眠13.5%(24/178例)、口渇10.7%(19/178例)、便秘10.1%(18/178例)、悪心9.6%(17/178例)、倦怠感6.7%(12/178例)、食欲減退5.1%(9/178例)であった22)。
17.1.10 国内第III相 長期投与試験
試験開始前の3ヵ月間で月に14日以上の痛みを有する特発性変形性膝関節症患者を対象として、本剤(デュロキセチンとして60mg)を最大50週間投与した結果、BPI-疼痛重症度(平均の痛み)スコアの変化量は表17-10のとおりであり、長期間にわたり鎮痛効果が維持された23)。
| 評価時期 | 例数 | BPI-疼痛重症度(平均の痛み)スコア | 変化量 |
| ベースライン | 93 | 3.04±1.76 | − |
| 8週時 | 91 | 1.80±1.33 | −1.25±1.35 |
| 16週時 | 88 | 1.59±1.34 | −1.44±1.38 |
| 28週時 | 87 | 1.43±1.24 | −1.63±1.42 |
| 50週時 | 81 | 1.52±1.54 | −1.53±1.41 |
副作用発現頻度は51.6%(48/93例)であった。主な副作用は便秘17.2%(16/93例)、傾眠12.9%(12/93例)、口渇11.8%(11/93例)であった23)。
18. 薬効薬理
19. 有効成分に関する理化学的知見
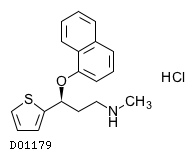
19.1. デュロキセチン塩酸塩
| 一般的名称 | デュロキセチン塩酸塩 |
|---|---|
| 一般的名称(欧名) | Duloxetine Hydrochloride |
| 化学名 | (+)-(S)-N-Methyl-3-(1-naphthyloxy)-3-(2-thienyl)propylamine monohydrochloride |
| 分子式 | C18H19NOS・HCl |
| 分子量 | 333.88 |
| 融点 | 165℃ |
| 物理化学的性状 | 白色の粉末又は塊である。 メタノール又はジメチルスルホキシドに溶けやすく、エタノール(99.5)にやや溶けやすく、水にやや溶けにくい。 |
| KEGG DRUG |
|
22. 包装
<サインバルタカプセル20mg>
500カプセル[瓶、バラ]
100カプセル[10カプセル(PTP)×10]
500カプセル[10カプセル(PTP)×50]
<サインバルタカプセル30mg>
500カプセル[瓶、バラ]
100カプセル[10カプセル(PTP)×10]
23. 主要文献
- 高橋明比古ほか, 臨床精神薬理, 12, 1411-1426, (2009)
- 高橋明比古ほか, 臨床精神薬理, 12, 1439-1454, (2009)
- 高橋明比古ほか, 臨床精神薬理, 12, 1455-1481, (2009)
- 熊谷雄治, 臨床精神薬理, 12, 1483-1497, (2009)
- 高橋明比古ほか, 臨床精神薬理, 12, 1427-1437, (2009)
- 社内資料:臨床における薬物動態(2010/1/20承認、申請資料概要2.7.1.2、2.7.2.2、2.7.2.3)
- 社内資料:ラットにおける胎盤・胎児移行性(2010/1/20承認、申請資料概要2.6.4.4)
- 社内資料:臨床における薬物相互作用試験(2010/1/20承認、申請資料概要2.7.2.2、2.7.6.3)
- 社内資料:デュロキセチンの酸化的代謝に関与するCYP分子種の同定(2010/1/20承認、申請資料概要2.6.4.7)
- 村崎光邦ほか, 臨床精神薬理, 12, 1499-1515, (2009)
- 樋口輝彦ほか, 臨床精神薬理, 12, 1613-1634, (2009)
- 樋口輝彦ほか, 臨床精神薬理, 12, 1579-1593, (2009)
- Yasuda H,et al., J Diabetes Investig., 2, 132-139, (2011) »PubMed »DOI
- Yasuda H,et al., J Diabetes Investig., 7, 100-108, (2016) »PubMed »DOI
- 社内資料:糖尿病性神経障害に伴う疼痛を対象とした国内第III相継続投与試験(2012/2/22承認、申請資料概要2.7.6.2)
- Murakami,M,et al., Arthritis Res Ther., 17, 224-236, (2015) »PubMed »DOI
- 社内資料:線維筋痛症を対象とした国内第III相プラセボ対照試験(2015/5/26承認、申請資料概要2.7.6.1)
- Murakami,M.et al., Mod Rheumatol., (2016), (doi:10.1080/14397595.2016.1245237) »PubMed »DOI
- Konno,S.,et al., Spine., 41, 1709-1717, (2016) »PubMed »DOI
- 社内資料:慢性腰痛症を対象とした国内第III相プラセボ対照試験(2016/3/18承認、申請資料概要2.7.6.1)
- 社内資料:慢性腰痛症を対象とした国内第III相継続長期投与試験(2016/3/18承認、申請資料概要2.7.6.5)
- 社内資料:変形性関節症を対象とした国内第III相プラセボ対照試験(2016/12/19承認、申請資料概要2.7.6.1)
- 社内資料:変形性関節症を対象とした国内第III相継続長期投与試験(2016/12/19承認、申請資料概要2.7.6.5)
- 社内資料:in vitro及びex vivoにおけるモノアミン取り込み阻害作用(2010/1/20承認、申請資料概要2.6.2.2)
- 社内資料:ラット及びマウスにおけるモノアミン取り込み阻害並びにin vitroにおけるモノアミン酸化酵素阻害作用(2010/1/20承認、申請資料概要2.6.2.2)
- 社内資料:脳内の細胞外モノアミン濃度増加作用(2010/1/20承認、申請資料概要2.6.2.2、2.6.2.6)
- 社内資料:脳内各種神経伝達物質受容体に対する特異性試験(2010/1/20承認、申請資料概要2.6.2.2)
- 社内資料:ラットにおける抗うつ作用(2010/1/20承認、申請資料概要2.6.2.2)
- 社内資料:神経障害性疼痛動物モデルにおける効果(2012/2/22承認、申請資料概要2.6.2.2)
- 社内資料:その他疼痛動物モデルにおける効果(2012/2/22承認、申請資料概要2.6.2.2)
24. 文献請求先及び問い合わせ先
文献請求先
製品情報問い合わせ先
26. 製造販売業者等
26.1 製造販売元
塩野義製薬株式会社
大阪市中央区道修町3丁目1番8号