医薬品情報
| 総称名 | ヴォリブリス |
|---|---|
| 一般名 | アンブリセンタン |
| 欧文一般名 | Ambrisentan |
| 製剤名 | アンブリセンタン錠 |
| 薬効分類名 | エンドセリン受容体拮抗薬 |
| 薬効分類番号 | 2190 |
| ATCコード | C02KX02 |
| KEGG DRUG |
D07077
アンブリセンタン
|
| JAPIC | 添付文書(PDF) |
添付文書情報2024年8月 改訂(第3版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ヴォリブリス錠2.5mg | Volibris Tablets2.5mg | グラクソ・スミスクライン | 2190031F1023 | 3080.1円/錠 | 処方箋医薬品 |
2. 禁忌
4. 効能または効果
5. 効能または効果に関連する注意
5.1 WHO機能分類クラスIVの患者における有効性及び安全性は確立していない。
5.2 本剤の使用にあたっては、最新の治療ガイドラインを参考に投与の要否を検討すること。
5.3 肺動脈性肺高血圧症の治療に十分な知識及び経験を有する医師のもとで、本剤の投与が適切と判断される患者に対して適用を考慮すること。
5.4 小児では、特発性又は遺伝性肺動脈性肺高血圧症、先天性心疾患の外科的修復術後の肺動脈性肺高血圧症及び結合組織疾患に伴う肺動脈性肺高血圧症以外の肺動脈性肺高血圧症における有効性・安全性は確立されていない。
6. 用法及び用量
成人
通常、成人にはアンブリセンタンとして5mgを1日1回経口投与する。なお、症状に応じて1日10mgを超えない範囲で適宜増量する。
小児
通常、8歳以上の小児には、体重に応じアンブリセンタンとして下記の投与量を1日1回経口投与する。
20〜35kg未満
通常、2.5mgとし症状に応じて1日5mgを超えない範囲で適宜増量する。
35〜50kg未満
通常、5mgとし症状に応じて1日7.5mgを超えない範囲で適宜増量する。
50kg以上
通常、5mgとし症状に応じて1日10mgを超えない範囲で適宜増量する。
7. 用法及び用量に関連する注意
8. 重要な基本的注意
8.1 エンドセリン受容体拮抗薬(ERA)の投与時に肝酵素上昇が認められているため、本剤の投与開始前に必ず肝機能検査を実施し、投与中においては必要に応じて定期的に、肝機能検査を実施しモニターすること。本剤投与中に、臨床的に顕著なアミノトランスフェラーゼ(AST、ALT)上昇、肝障害の徴候を伴うアミノトランスフェラーゼ上昇、又は黄疸が発現した場合には本剤の投与を中止すること。
8.2 ヘモグロビン減少及びヘマトクリット減少が起こる可能性があり、貧血に至った症例があるため、投与開始前及び投与開始1ヵ月後に血液検査を実施すること。また、その後も定期的に検査を実施することが望ましい。[9.1.1、11.1.1参照]
8.3 本剤の投与により急性肺水腫の徴候が見られた場合は、肺静脈閉塞性疾患の可能性を考慮すること。[9.1.3参照]
8.4 特発性肺線維症(IPF)を対象とした海外臨床試験において、本剤投与によりIPFの病態増悪リスクの増加の可能性が示されている。肺の線維化を伴う肺動脈性肺高血圧症の患者に本剤を投与する際は、肺線維症の治療に精通した呼吸器科医に相談するなど、本剤投与によるリスクとベネフィットを考慮した上で、投与の可否を慎重に検討すること。[15.1参照]
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 重度の貧血患者
9.1.2 間質性肺炎患者
間質性肺炎が増悪することがある。[11.1.4参照]
9.1.3 肺静脈閉塞性疾患を有する患者
本剤を投与しないことが望ましい。心血管系の状態を著しく悪化させるおそれがある。[8.3参照]
9.1.4 出血の危険因子を有する患者
出血の危険性に注意すること。国内臨床試験において鼻出血など出血の副作用が認められている。
9.2 腎機能障害患者
9.2.1 重度の腎障害のある患者
これらの患者を対象とした臨床試験は実施していない。
9.3 肝機能障害患者
9.3.1 重度の肝障害のある患者
9.3.2 中等度の肝障害のある患者
本剤の血中濃度が上昇するおそれがある。[16.6.2参照]
9.3.3 投与開始前のアミノトランスフェラーゼ(AST、ALT)のいずれかが基準値上限の3倍を超える患者
肝機能障害を増悪させるおそれがある。[16.6.2参照]
9.4 生殖能を有する者
本剤の投与に際し、妊娠する可能性のある女性には以下について説明すること。また、必要に応じて投与前又は投与期間中に定期的に妊娠検査を行うこと。[9.5参照]
・妊娠中に本剤を服用した場合の胎児に及ぼす危険性。
・本剤の投与中及び最終投与後5日間において避妊する必要性及び適切な避妊法。
・妊娠した場合若しくはその疑いがある場合には、医師に直ちに連絡すること。
9.5 妊婦
9.6 授乳婦
本剤投与中は授乳しないことが望ましい。母動物(ラット)に妊娠15日から分娩後20日まで経口投与した結果、出生児生存率の低下が認められている。
9.7 小児等
低出生体重児、新生児、乳児、幼児、8歳未満又は体重20kg未満の小児を対象とした臨床試験は実施していない。
9.8 高齢者
一般に、生理機能が低下していることが多い。海外臨床試験において、末梢性浮腫の多くは軽度から中等度であったが、高齢者では発現する可能性が高く、重症例が多い傾向が示唆された。
10. 相互作用
10.2 併用注意
| シクロスポリン [7.、16.7.4参照] | シクロスポリンとの併用により本剤のAUCが約2倍になるとの報告がある。併用する場合には、本剤は成人及び50kg以上の小児は1日1回5mg、50kg未満の小児は1日1回2.5mgを上限として投与すること。 | 詳細な機序は不明であるが、シクロスポリンとの併用により、本剤の血中濃度が上昇する。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 貧血(7.6%)
11.1.2 体液貯留(頻度不明)
異常が認められた場合には本剤に起因するものか、基礎疾患の心不全によるものか原因を確認し、本剤の投与中止、利尿剤の投与などの処置を行うこと。
11.1.3 心不全(1.5%)
体液貯留に関連し、心不全があらわれることがある。
11.1.4 間質性肺炎(頻度不明)
間質性肺炎が発現又は増悪することがある。咳嗽、呼吸困難、発熱、肺音の異常(捻髪音)等が認められた場合には、速やかに胸部X線、胸部CT、血清マーカー等の検査を実施すること。異常が認められた場合には、投与を中止し、副腎皮質ホルモン剤の投与等の適切な処置を行うこと。[9.1.2参照]
11.2 その他の副作用
| 10%以上 | 10%未満 | 頻度不明 | |
| 過敏症 | 過敏症反応(血管性浮腫、発疹等) | ||
| 精神神経系 | 頭痛 | めまい | |
| 循環器 | 潮紅 | 動悸、低血圧 | |
| 呼吸器 | 鼻閉注1) | 鼻出血、喀血、呼吸困難注2)、副鼻腔炎、鼻咽頭炎 | |
| 消化器 | 便秘、悪心、腹痛、嘔吐 | ||
| 肝臓 | トランスアミナーゼ上昇 | ||
| 全身症状 | 末梢性浮腫 | 疲労 | 無力症 |
| 眼 | 視覚障害(霧視等)、眼窩周囲浮腫 | ||
| 血液 | 白血球減少 |
13. 過量投与
13.1 症状
本剤50mg及び100mg(推奨最高用量の5倍から10倍)を健康成人に単回投与したところ、本剤との関連性が否定できない頭痛、潮紅、浮動性めまい、悪心及び鼻閉が発現した。
14. 適用上の注意
14.1 薬剤交付時の注意
14.1.1 PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
14.1.2 PTPシートからの取り出しは、裏のラベルを剥がした後、指の腹で押し出すこと。
15. その他の注意
15.1 臨床使用に基づく情報
適応外であるが、海外で実施された特発性肺線維症(IPF)患者492例(うち二次性肺高血圧症患者54例)を対象としたプラセボ対照臨床試験の中間解析の結果、IPFの病態の悪化(呼吸器系の障害による入院を含む)又は死亡がプラセボ群と比較して本剤投与群で多くみられ(本剤投与群329例中90例(27%)、プラセボ群163例中28例(17%))、試験が中止された。[8.4参照]
15.2 非臨床試験に基づく情報
15.2.1 ラットでは精細管萎縮、精子形態異常、精子数減少、交尾率及び受胎率の低値が、イヌでも精細管萎縮、空胞化、拡張などが認められている。なお、ヒトの男性生殖能に対する影響は不明である。
15.2.2 ヒト末梢リンパ球を用いる染色体異常試験では高濃度で染色体の構造異常がみられたが、細菌を用いる復帰突然変異試験、ラットを用いる小核試験及び肝不定期DNA合成試験の結果は陰性であった。
15.2.3 ラットでは鼻腔の炎症及び鼻甲介骨過形成がみられ、イヌでは炎症のみが認められている。
15.2.4 幼若ラットの反復投与毒性試験において、生後7日から生後26日、36日又は62日まで1日1回経口投与した結果、異常呼吸音、無呼吸、低酸素血症及び脳重量の低値が認められている(ヒト小児推定AUCの2.3〜3.8倍)。なお、脳の病理組織学的変化は認められていない。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
健康成人男性に本剤2.5mg注)、5mg又は10mgを単回経口投与した時、本剤は速やかに吸収され、投与後2〜2.5時間(中央値)に最高血漿中濃度(Cmax)に達した。Cmax及び血漿中濃度−時間曲線下面積(AUC)は用量の増加にほぼ比例して増加した。消失半減期(t1/2)は約10〜19時間であった。
図1 空腹時単回投与後の血漿中アンブリセンタン濃度の推移(平均値±標準偏差)
| 投与量(例数) | Cmax(ng/mL) | tmax(h) | AUC0-∞(ng・h/mL) | t1/2(h) |
| 2.5mg注)(11例) | 178.7±32.05 | 2.5(1.0-4.0) | 1438.8±372.60 | 10.0±3.62 |
| 5mg(11例) | 362.0±42.53 | 2.0(1.0-4.0) | 2944.5±608.55 | 13.6±4.83 |
| 10mg(12例) | 766.8±90.68 | 2.0(1.0-4.0) | 6894.1±1612.50 | 18.8±10.98 |
注)本剤の成人承認用量は1日1回5mg、症状に応じて1日10mgを超えない範囲で適宜増量である。
16.1.2 反復投与
成人肺動脈性肺高血圧症(PAH)患者に本剤5mgを1日1回12週間反復経口投与した時、投与後4時間にCmaxに達し、t1/2は11時間であった。定常状態におけるAUC0-24は8337.4ng・h/mL、Cmaxは674.3ng/mLであった。
また、本剤5mg及び10mgを投与した時の定常状態時における投与前及び投与後2〜4時間の血漿中アンブリセンタン濃度は表2のとおりであった。
また、本剤5mg及び10mgを投与した時の定常状態時における投与前及び投与後2〜4時間の血漿中アンブリセンタン濃度は表2のとおりであった。
| 投与群(症例数) | 血漿中アンブリセンタン濃度(ng/mL):投与前 | 血漿中アンブリセンタン濃度(ng/mL):投与2〜4時間後 |
| 5mg(28例) | 147.8±157.2 | 635.2±260.7 |
| 10mg(17例) | 263.3±265.5 | 1083.2±318.9 |
8歳以上の小児PAH患者に本剤を1日1回経口投与した時の用量群別の曝露量(推定値)は表3のとおりであった。
| 集団 | 投与量 (症例数) | 体重区分 | AUCss(μg・h/mL) | Cmax,ss(ng/mL) |
| 低用量群 | ||||
| 日本人 | 2.5mg (3例) | 20〜35kg未満 | 3.94(3.03-5.11) | 442(217-898) |
| 5mg (1例) | 35〜50kg未満 | 5.60 | 668 | |
| 5mg (1例) | 50kg以上 | 3.87 | 530 | |
| 外国人 | 2.5mg (5例) | 20〜35kg未満 | 4.09(3.11-5.39) | 501(407-615) |
| 5mg (7例) | 35〜50kg未満 | 6.56(5.00-8.62) | 497(384-643) | |
| 5mg (3例) | 50kg以上 | 3.84(1.98-7.45) | 659(255-1700) | |
| 高用量群 | ||||
| 外国人 | 5mg (9例) | 20〜35kg未満 | 8.73(7.75-9.83) | 953(850-1070) |
| 7.5mg (4例) | 35〜50kg未満 | 8.70(7.62-9.92) | 1100(679-1790) | |
| 10mg (6例) | 50kg以上 | 10.2(8.04-12.8) | 948(791-1140) | |
16.1.3 母集団薬物動態解析
健康成人及び成人PAH患者における母集団薬物動態解析の結果から、年齢及び性別は本剤の薬物動態に大きな影響を与えなかった(外国人データ)。
16.2 吸収
16.2.1 食事の影響
健康成人男性に本剤10mgを空腹時又は食後(標準的な朝食)単回経口投与した時、食後投与では空腹時投与と比較し、Cmaxは約17%低下したが、AUC0-48、最高血漿中濃度到達時間(tmax)及びt1/2には影響は認められなかった。
| 投与量(例数) | Cmax(ng/mL) | tmax(h) | AUC0-48(ng・h/mL) | t1/2(h) |
| 10mg(12例)空腹時 | 766.8±90.68 | 2.0(1.0-4.0) | 6437.3±1487.68 | 18.8±10.98 |
| 10mg(12例)食後 | 637.1±102.65 | 2.5(1.5-4.0) | 6251.9±1389.96 | 19.9±11.20 |
16.3 分布
In vitroでの本剤(0.2〜20μg/mL)のヒト血漿蛋白結合率は98.8%であった。また、本剤は主にアルブミンと結合し(96.5%)、一部はα1-酸性糖蛋白質と結合した。
16.4 代謝
本剤はin vitroでUDP-グルクロン酸転移酵素のUGT1A9、UGT2B7及びUGT1A3によりグルクロン酸抱合され、その他に、チトクロームP450(CYP)で酸化的に代謝される。CYPによる代謝には主にCYP3A4、一部にCYP2C19及びCYP3A5が関与する。
16.5 排泄
健康成人男性を対象に2H及び14C標識した本剤を単回経口投与した時の主要排泄経路は糞中であり、投与量の約40%が未変化体、約21%が4-水酸化体として糞中に排泄された。また、尿中には、投与量の約4%が未変化体、約18%が未変化体のグルクロン酸抱合体及び4-水酸化体のグルクロン酸抱合体として排泄された(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎障害患者
腎障害患者における本剤の薬物動態は検討されていない。
本剤の主要排泄経路は糞中であるため、腎障害患者では、本剤の血中濃度が上昇する可能性は低い。
本剤の主要排泄経路は糞中であるため、腎障害患者では、本剤の血中濃度が上昇する可能性は低い。
16.6.2 肝障害患者
16.7 薬物相互作用
16.7.1 代謝酵素に及ぼす影響
非臨床試験において、本剤は第I及びII相代謝酵素を阻害・誘導しなかったことから、本剤がこれらの代謝酵素で代謝される薬剤の体内動態に影響を及ぼす可能性は低いと考えられる。
16.7.2 薬剤トランスポーターに及ぼす影響
本剤はin vitroでP-糖蛋白質及びorganic anion transporting polypeptide(OATP)の基質である。また、本剤はin vitroでOATP1B1、OATP1B3及びsodium taurocholate co-transporting polypeptide(NTCP)を阻害し、IC50はそれぞれ47、45及び約100μMであった。本剤はin vitroでP-糖蛋白質、bile salt export pump(BSEP)、breast cancer resistance protein(BCRP)及びmulti-drug resistance protein-2(MRP2)を阻害しなかった。
16.7.3 CYP3A4に対する誘導の検討
健康成人を対象に本剤がCYP3A4を誘導する可能性について尿中6β-ヒドロキシコルチゾール濃度を指標として検討した結果、本剤はCYP3A4を誘導しなかった(外国人データ)。
16.7.4 シクロスポリン
健康成人男女に、本剤5mg反復投与時にシクロスポリン100〜150mgを併用した結果、定常状態における本剤のAUCは約2倍となった。シクロスポリン100〜150mgを反復投与時に本剤5mgを併用した結果、本剤は定常状態におけるシクロスポリンの薬物動態に影響を与えなかった(外国人データ)。[7.、10.2参照]
16.7.5 ケトコナゾール(経口剤:国内未発売)
健康成人男性に、ケトコナゾール400mg反復投与時に本剤10mgを併用した結果、本剤のCmax及びAUCは非併用時に比べ、それぞれ約20%及び35%増加した(外国人データ)。
16.7.6 リファンピシン
健康成人男女に、本剤10mg反復投与時にリファンピシン600mgを併用した結果、リファンピシン併用初期には本剤のAUCの一過性の増加(約2倍)が認められたが、リファンピシンを8日間併用投与後には、リファンピシンは本剤の薬物動態に影響を与えなかった(外国人データ)。
16.7.7 経口避妊薬(エチニルエストラジオール35μg及びノルエチステロン1mg含有)
健康成人女性に、本剤10mg反復投与時に経口避妊薬を併用した結果、本剤はエチニルエストラジオール及びノルエチステロンの薬物動態に影響を与えなかった(外国人データ)。
16.7.8 ジゴキシン
健康成人男性に、本剤10mg反復投与時にジゴキシン0.5mgを併用した結果、本剤はジゴキシンの薬物動態に影響を与えなかった(外国人データ)。
16.7.9 オメプラゾール
オメプラゾールによる血漿中未変化体濃度及び薬物動態に与える影響を評価するため、PAH患者での長期第III相試験における薬物動態データを用いてpost-hoc解析を行ったところ、オメプラゾール併用投与群と非併用投与群で差は認められなかった(外国人データ)。
16.7.10 その他の薬剤
健康成人男女に、本剤10mgとシルデナフィル20mg、タダラフィル40mg、又はワルファリン25mgを併用した結果、本剤の薬物動態に変化は認められなかった。また、本剤はシルデナフィル、タダラフィル、ワルファリンの薬物動態に影響を与えなかった(外国人データ)。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国内第II/III相試験(成人)
PAH患者を対象としたオープンラベル、非対照、用量漸増試験において、本剤5mgを1日1回12週間、その後用量調節期間として本剤5〜10mgを12週間投与した結果、投与12週時及び24週時の6分間歩行距離(6MWD:主要評価項目)、ボルグ呼吸困難指数(BDI)、WHOの肺高血圧症機能分類(WHO機能分類)及び血漿中脳性ナトリウム利尿ペプチド(血漿中BNP)濃度がベースラインから改善し、24週間の投与期間中にPAHの臨床的な増悪を認めた被験者は1例であった(表1)。さらに、投与12週時及び24週時の血行動態の改善も認められた(表2)1)。
副作用発現頻度は、80%(20/25例)であった。主な副作用は、頭痛36%(9/25例)、鼻閉20%(5/25例)、ほてり16%(4/25例)、潮紅12%(3/25例)、末梢性浮腫、発疹、血圧低下、浮動性めまい、鼻出血、貧血各8%(各2/25例)であった。
副作用発現頻度は、80%(20/25例)であった。主な副作用は、頭痛36%(9/25例)、鼻閉20%(5/25例)、ほてり16%(4/25例)、潮紅12%(3/25例)、末梢性浮腫、発疹、血圧低下、浮動性めまい、鼻出血、貧血各8%(各2/25例)であった。
| 時期 | 投与12週 | 投与24週 |
| 症例数 | 25例 | 25例 |
| 6MWDの変化量,m,平均値±SD | 33.49±43.24 | 46.82±52.71 |
| BDIの変化量,平均値±SD | −0.60±2.16 | −0.69±1.90 |
| WHO機能分類の変化,症例数(%) | ||
| 改善 | 9(36) | 10(40) |
| 変化なし | 16(64) | 14(56) |
| 悪化 | 0 | 1(4) |
| PAHの臨床的な増悪注1)を認めた被験者数(%) | 0 | 1(4) |
| BNPの変化量,ng/L,平均値±SD | −76.86±160.94 | −60.15±248.35 |
| 時期 | 投与12週 | 投与24週 |
| 症例数(25例) | 21例 | 16例 |
| 血行動態の変化,平均値±SD | ||
| 平均肺動脈圧(mPAP),mmHg | −6.29±11.20 | −8.69±13.90 |
| 平均右房圧(mRAP),mmHg | −1.12±3.76 | −0.69±3.68 |
| 心係数(CI)注1),L/min/m2 | 0.67±0.58 | 0.63±0.62 |
| 肺血管抵抗(PVR),mmHg/L/min | −7.26±7.43 | −8.35±7.64 |
17.1.2 国内第III相試験(成人)
PAH患者を対象に本剤5〜10mgを投与した多施設共同、オープンラベル、用量漸増、延長試験(平均投与期間:138.6週間、最長投与期間:164.1週間)でも本剤の改善効果(6MWD、WHO機能分類、BDI、BNPの改善)が維持された。本試験期間中にPAHの臨床的な増悪を認めた被験者は1例であった2)。
副作用発現頻度は、43%(9/21例)であった。主な副作用は、喀血14%(3/21例)、鼻出血、ほてり各10%(各2/21例)であった(承認申請時の中間解析結果)。
副作用発現頻度は、43%(9/21例)であった。主な副作用は、喀血14%(3/21例)、鼻出血、ほてり各10%(各2/21例)であった(承認申請時の中間解析結果)。
17.1.3 海外第II相試験(成人)
PAH患者を対象に、本剤1mg注)、2.5mg注)、5mg又は10mgを1日1回12週間盲検下で投与後注)、12週間非盲検下で本剤を投与した用量設定の第II相試験を実施した結果、6MWD(主要評価項目)、BDI、WHO機能分類、被験者の概括評価(QOL)及び血行動態の改善が認められた3)。
副作用発現頻度は、本剤併合群(1mg、2.5mg、5mg、10mg)注)で59.4%(38/64例)であった。主な副作用は、鼻閉20.3%(13/64例)、末梢性浮腫15.6%(10/64例)、頭痛14.1%(9/64例)、悪心、潮紅、ALT増加各10.9%(各7/64例)などであった。
副作用発現頻度は、本剤併合群(1mg、2.5mg、5mg、10mg)注)で59.4%(38/64例)であった。主な副作用は、鼻閉20.3%(13/64例)、末梢性浮腫15.6%(10/64例)、頭痛14.1%(9/64例)、悪心、潮紅、ALT増加各10.9%(各7/64例)などであった。
17.1.4 海外第II相試験(成人)
血清アミノトランスフェラーゼ異常のため、過去に本剤以外のERA(ボセンタン、sitaxentan又は両剤)の投与を中止したPAH患者を対象とした非盲検の第II相試験を実施した。本試験の主目的は、血清アミノトランスフェラーゼ異常のために過去にERAの投与を中止した被験者における血清アミノトランスフェラーゼ異常の発現頻度の評価であったが、有効性の評価項目のデータも得られている。本試験で投与12週後に基準値上限の3倍を超える血清アミノトランスフェラーゼ異常が認められた被験者は1例であり、本被験者では本剤の投与が一時中断された。また、本剤投与により6MWD、BDI、WHO機能分類、QOL(SF-36)の改善が認められた4)。
副作用発現頻度は、55.6%(20/36例)であった。2例以上に発現した副作用は、頭痛、潮紅各13.9%(各5/36例)、末梢性浮腫11.1%(4/36例)、体液貯留8.3%(3/36例)であった(承認申請時の中間解析結果)。
副作用発現頻度は、55.6%(20/36例)であった。2例以上に発現した副作用は、頭痛、潮紅各13.9%(各5/36例)、末梢性浮腫11.1%(4/36例)、体液貯留8.3%(3/36例)であった(承認申請時の中間解析結果)。
17.1.5 海外第III相試験(成人)
PAH患者を対象に、本剤2.5mg注)、5mg又は10mgを12週間盲検下で投与した同一デザインのプラセボ対照の第III相試験を2試験実施して併合解析した結果、本剤投与群ではプラセボ群に比べて主要評価項目の6MWDの有意な改善が認められた。また、本剤併合群ではプラセボ群に比べて他の副次評価項目の有意な改善が認められ、血漿中BNP濃度も有意に低下した(表3)。さらに、本剤併合群ではプラセボ群に比べて副次評価項目であるPAHの臨床的な増悪を認めるまでの時間が有意に遅延した(図1)。
副作用発現頻度は、本剤併合群(2.5mg、5mg、10mg)注)で39.5%(103/261例)であった。主な副作用は、頭痛9.6%(25/261例)、末梢性浮腫9.2%(24/261例)、鼻閉3.8%(10/261例)であった。
副作用発現頻度は、本剤併合群(2.5mg、5mg、10mg)注)で39.5%(103/261例)であった。主な副作用は、頭痛9.6%(25/261例)、末梢性浮腫9.2%(24/261例)、鼻閉3.8%(10/261例)であった。
| 投与群 | プラセボ | 2.5mg | 5mg | 10mg | 本剤併合 |
| 症例数 | 132例 | 64例 | 130例 | 67例 | 261例 |
| 6MWDの変化量,m,平均値±SD | −9.0±86.22 | 22.2±82.67 | 35.7±80.18 | 43.6±65.91 | 34.4±77.51 |
| BDIの変化量,平均値±SD | 0.40±2.46 | −0.20±2.17 | −0.34±1.96 | −0.88±1.93 | −0.45±2.01 |
| WHO機能分類の変化,症例数(%) | |||||
| 改善 | 27(20.5) | 10(15.6) | 28(21.5) | 20(29.9) | 58(22.2) |
| 変化なし | 82(62.1) | 51(79.7) | 99(76.2) | 44(65.7) | 194(74.3) |
| 悪化 | 23(17.4) | 3(4.7) | 3(2.3) | 3(4.5) | 9(3.4) |
| QOL(SF-36の身体機能),平均値±SD | 1.07±7.64 | 3.86±7.14 | 3.34±8.30 | 4.52±7.16 | 3.77±7.73 |
| PAHの臨床的な増悪注1)を認めた被験者数(%) | 20(15.2) | 3(4.7) | 6(4.6) | 3(4.5) | 12(4.6) |
| BNPの変化量,ng/L,平均値±SD | 29.17±231.19 | −98.64±195.42 | −90.63±304.66 | −149.32±226.32 | −106.99±262.45 |
図1 PAHの臨床的な増悪を認めるまでの時間のカプランマイヤー曲線
17.1.6 海外第II相試験(成人)
用量設定の第II相試験に参加したPAH患者は、その後長期投与試験に移行し、継続して有効性の各評価項目を検討した結果、本剤の改善効果(6MWD、WHO機能分類、BDIの改善)は約3年間おおむね維持された。
また、PAH患者の生存期間を評価した結果、本剤投与1年後の生存率が93%、投与2年後の生存率が87%、投与3年後の生存率が85%であった。
副作用発現頻度は、本剤併合群(1mg、2.5mg、5mg、10mg)注)で53.7%(29/54例)であった。5%以上に発現した副作用は、鼻閉14.8%(8/54例)、末梢性浮腫、頭痛各7.4%(各4/54例)、浮動性めまい5.6%(3/54例)であった(承認申請時の中間解析結果)。
また、PAH患者の生存期間を評価した結果、本剤投与1年後の生存率が93%、投与2年後の生存率が87%、投与3年後の生存率が85%であった。
副作用発現頻度は、本剤併合群(1mg、2.5mg、5mg、10mg)注)で53.7%(29/54例)であった。5%以上に発現した副作用は、鼻閉14.8%(8/54例)、末梢性浮腫、頭痛各7.4%(各4/54例)、浮動性めまい5.6%(3/54例)であった(承認申請時の中間解析結果)。
17.1.7 海外第III相試験(成人)
プラセボ対照の第III相試験に参加したPAH患者は、その後長期投与試験に移行し、継続して有効性の各評価項目を検討した結果、本剤の改善効果(6MWD、WHO機能分類、BDIの改善)は少なくとも3年間維持された。
また、PAH患者の生存期間を評価した結果、本剤投与1年後の生存率が約93%、投与2年後の生存率が約85%、投与3年後の生存率が約79%であり、本剤の長期投与によりPAH患者の生存率は高いまま維持されることが示された。
副作用発現頻度は、本剤併合群(2.5mg、5mg、10mg)注)で44.6%(171/383例)であった。10%以上に発現した副作用は、末梢性浮腫11.8%(45/383例)、頭痛10.2%(39/383例)であった(承認申請時の中間解析結果)。
また、PAH患者の生存期間を評価した結果、本剤投与1年後の生存率が約93%、投与2年後の生存率が約85%、投与3年後の生存率が約79%であり、本剤の長期投与によりPAH患者の生存率は高いまま維持されることが示された。
副作用発現頻度は、本剤併合群(2.5mg、5mg、10mg)注)で44.6%(171/383例)であった。10%以上に発現した副作用は、末梢性浮腫11.8%(45/383例)、頭痛10.2%(39/383例)であった(承認申請時の中間解析結果)。
注)本剤の成人承認用量は1日1回5mg、症状に応じて1日10mgを超えない範囲で適宜増量である。
17.1.8 国際共同第II相試験(小児)
8歳以上18歳未満のPAH患者を対象としたオープンラベル試験において、アンブリセンタンの2用量群(低用量群及び高用量群)のいずれかに無作為化し、各被験者の体重区分に応じた用量にて本剤を1日1回24週間投与し、安全性及び有効性を評価した。投与開始前のWHO機能分類の内訳は、クラスIIが32例、クラスIIIが9例であった。また、肺動脈性肺高血圧症の臨床分類の内訳は、特発性PAHが26例、先天性心疾患の外科的修復術後も持続するPAHが11例、家族性PAHが3例、結合組織病に伴うPAHが1例であった。低用量群及び高用量群ともに体重20kg以上35kg未満の患者には本剤2.5mgの用量で投与を開始し、高用量群では2週間後に5mgに増量した。体重35kg以上の患者には本剤5mgの用量で投与を開始し、高用量群は体重35kg以上50kg未満の場合は7.5mgに、体重50kg以上の場合は10mgにそれぞれ2週間後に増量した。評価例数41例(日本人症例5例含む)において、6MWDにベースラインから改善が認められ、WHO機能分類のベースラインからの変化は、表4のとおりであった。また、血行動態のベースラインからの変化は、表5のとおりであった。
副作用発現頻度は、低用量群及び高用量群それぞれで、38%(8/21例)及び35%(7/20例)であった。主な副作用は、低用量群では頭痛14%(3/21例)、末梢性浮腫10%(2/21例)、鼻閉10%(2/21例)であり、高用量群では頭痛15%(3/20例)、悪心10%(2/20例)であった。
副作用発現頻度は、低用量群及び高用量群それぞれで、38%(8/21例)及び35%(7/20例)であった。主な副作用は、低用量群では頭痛14%(3/21例)、末梢性浮腫10%(2/21例)、鼻閉10%(2/21例)であり、高用量群では頭痛15%(3/20例)、悪心10%(2/20例)であった。
| 用量群 | 低用量群 | 高用量群 |
| 症例数 | 21例 | 20例 |
| 6MWDの変化量,m,平均値±SD | 55.14±102.182 | 26.25±62.011 |
| WHO機能分類の変化,症例数(%) | ||
| 改善 | 6(32) | 4(22) |
| 変化なし | 12(63) | 14(78) |
| 悪化 | 1(5) | 0 |
| PAHの臨床的な増悪注1)を認めた被験者数(%) | 3(14) | 3(15) |
| 症例数 | 5例注1) |
| 血行動態の変化,平均値±SD | |
| mPAP,mmHg | −2.2±6.06 |
| RAP,mmHg | 1.4±2.88 |
| CI,L/min/m2 | 0.94±0.658 |
| PVR,mmHg/L/min | −3.46±1.903 |
| 肺血管抵抗係数(PVRI),mmHg/L/min/m2 注2) | −3.589±2.5998 |
17.1.9 国際共同第II相長期継続投与試験(小児)
8歳以上18歳未満のPAH患者を対象としたオープンラベル試験に参加したPAH患者を対象に本剤2.5mg、5mg、7.5mg又は10mgのいずれかの用量(0.25mg/kg/日を超えない範囲で患者により調整)を1日1回投与した継続試験(先行試験を含む平均投与期間:1238.5日、最長投与期間:2346日)で長期の安全性及び有効性を評価した。評価例数38例(日本人症例5例含む)において、先行試験で認められた本剤の効果(6MWD、WHO機能分類)が試験期間を通して維持された。本継続投与試験中にPAHの臨床的な増悪は11例に報告され、初回のPAHの臨床的な増悪を認めるまでの時間は791.5±650.1日(平均値±SD)であった。本試験の中間解析時までに本試験を完了又は中止した34例の本剤の最終用量の内訳は2.5mgが4例、5mgが16例、7.5mgが4例、10mgが10例であった。
副作用発現頻度は、全体で39%(15/38例)であった。主な副作用は、頭痛8%(3/38例)、貧血5%(2/38例)、胃腸炎5%(2/38例)であった(承認申請時の中間解析結果)。
副作用発現頻度は、全体で39%(15/38例)であった。主な副作用は、頭痛8%(3/38例)、貧血5%(2/38例)、胃腸炎5%(2/38例)であった(承認申請時の中間解析結果)。
18. 薬効薬理
18.1 作用機序
本剤はエンドセリン(ET)受容体のうちETA受容体に高親和性、ETB受容体には低親和性(ETA受容体に比べて1/4000以下の親和性)を示す選択的ETA受容体拮抗薬である。PAH患者において血漿中ET-1濃度は高く、右心房圧や病態の程度と相関することなどから、ET-1がPAHの発症及び進展に重要であると考えられている5)。本剤は、肺血管ETA受容体阻害作用を介して内因性のET-1による肺血管平滑筋の収縮及び増殖を抑制し、PAHの症状を改善すると考えられる。
18.2 肺高血圧症モデルにおける作用
モノクロタリン誘発肺高血圧症モデルラットにおいて、4週間の反復経口投与により肺高血圧症の症状(右心室収縮期圧の上昇、右心肥大及び肺血管中膜肥厚)をそれぞれ有意に抑制した6)。
19. 有効成分に関する理化学的知見
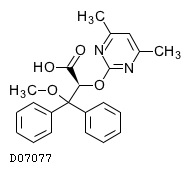
19.1. アンブリセンタン
| 一般的名称 | アンブリセンタン |
|---|---|
| 一般的名称(欧名) | Ambrisentan |
| 化学名 | (2S)-2-[(4,6-Dimethylpyrimidin-2-yl)oxy]-3-methoxy-3,3-diphenylpropanoic acid |
| 分子式 | C22H22N2O4 |
| 分子量 | 378.42 |
| 物理化学的性状 | 白色の結晶性の粉末である。 |
| 分配係数 | (logP):1.20(1-オクタノール/水) |
| KEGG DRUG |
|
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
60錠[10錠(PTP)×6]
23. 主要文献
- Yoshida S,et al., Curr Med Res Opin., 27, 1827-1834, (2011) »PubMed »DOI
- Yoshida S,et al., Curr Med Res Opin., 28, 1069-1076, (2012) »PubMed »DOI
- Galie N,et al., J Am Coll Cardiol., 46, 529-535, (2005) »PubMed »DOI
- McGoon MD,et al., Chest., 135, 122-129, (2009) »PubMed »DOI
- Galie N,et al., Cardiovasc Res., 61, 227-237, (2004) »PubMed »DOI
- Schroll S,et al., Scand J Clin Lab Invest., 68, 270-276, (2008) »PubMed »DOI
24. 文献請求先及び問い合わせ先
文献請求先
グラクソ・スミスクライン株式会社
カスタマー・ケア・センター
東京都港区赤坂1-8-1
電話:0120-561-007 (9:00〜17:45/土日祝日及び当社休業日を除く)
製品情報問い合わせ先
グラクソ・スミスクライン株式会社
カスタマー・ケア・センター
東京都港区赤坂1-8-1
電話:0120-561-007 (9:00〜17:45/土日祝日及び当社休業日を除く)
26. 製造販売業者等
26.1 製造販売元
グラクソ・スミスクライン株式会社
東京都港区赤坂1-8-1