医薬品情報
| 総称名 | レグナイト |
|---|---|
| 一般名 | ガバペンチン エナカルビル |
| 欧文一般名 | Gabapentin Enacarbil |
| 製剤名 | ガバペンチン エナカルビル錠 |
| 薬効分類名 | レストレスレッグス症候群治療剤 |
| 薬効分類番号 | 1190 |
| KEGG DRUG |
D09539
ガバペンチンエナカルビル
|
| KEGG DGROUP |
DG01245
ガバペンチン
|
| JAPIC | 添付文書(PDF) |
添付文書情報2020年11月 改訂(第1版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| レグナイト錠300mg | Regnite Tablets 300mg | アステラス製薬 | 1190020F1020 | 46.7円/錠 | 処方箋医薬品 |
2. 禁忌
4. 効能または効果
中等度から高度の特発性レストレスレッグス症候群(下肢静止不能症候群)
5. 効能または効果に関連する注意
5.1 レストレスレッグス症候群(下肢静止不能症候群)の診断は、国際レストレスレッグス症候群研究グループの診断基準及び重症度スケール(IRLS(International Restless Legs Syndrome Rating Scale))に基づき慎重に実施し、基準を満たす場合にのみ投与すること。
5.2 本剤は、原則、ドパミンアゴニストによる治療で十分な効果が得られない場合、又はオーグメンテーション(症状発現が2時間以上早まる、症状の増悪、他の部位への症状拡大)等によりドパミンアゴニストが使用できない場合に限り投与すること。国内臨床試験において主要評価項目である治療期最終時点におけるIRLS合計スコアの変化量ではプラセボ群との差は認められていない。[17.2.1参照]
6. 用法及び用量
通常、成人にはガバペンチン エナカルビルとして1日1回600mgを夕食後に経口投与する。
7. 用法及び用量に関連する注意
8. 重要な基本的注意
8.1 本剤の投与により体重増加を来すことがあるので、肥満に注意し、肥満の徴候があらわれた場合は、食事療法、運動療法等の適切な処置を行うこと。特に、投与量の増加、あるいは長期投与に伴い体重増加が認められることがあるため、定期的に体重計測を実施すること。
8.2 眠気、注意力・集中力・反射運動能力等の低下が起こることがあるので、本剤投与中の患者には自動車の運転等、危険を伴う機械の操作に従事させないよう注意すること。
8.3 本剤の投与により、霧視、調節障害等の眼障害が生じる可能性があるので、診察時に、眼障害について問診を行う等注意し、異常が認められた場合には適切な処置を行うこと。[15.2.1参照]
8.4 効果が認められない場合には、漫然と投与しないよう注意すること。
9. 特定の背景を有する患者に関する注意
9.2 腎機能障害患者
9.2.1 高度の腎機能障害患者(クレアチニンクリアランス30mL/min未満)
9.5 妊婦
妊婦又は妊娠している可能性のある女性には治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。妊娠ラットで胎盤及び胎児へ移行することが報告されている。さらに、妊娠ラット及び妊娠ウサギに投与した際に母動物に体重減少等がみられ、非妊娠動物に投与した場合と比較して毒性が増強する可能性が報告されている。また、早産あるいは流産(ウサギ)、胎児の低体重(ラット及びウサギ)、新生児の生存率低下及び低体重(ラット)が認められている。
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。動物実験(ラット)で乳汁中へ移行することが報告されている。
9.7 小児等
小児等を対象とした有効性及び安全性を指標とした臨床試験は実施していない。
9.8 高齢者
10. 相互作用
10.2 併用注意
| モルヒネ | 本剤の活性代謝物であるガバペンチンの併用によりガバペンチンのCmaxが24%、AUCが44%それぞれ増加したとの報告がある。本剤併用時にもガバペンチンの血中濃度が上昇するおそれがあるので、傾眠等の中枢神経抑制症状に注意し、必要に応じて本剤又はモルヒネの用量を減量すること。 | 機序は不明だが、モルヒネにより消化管運動が抑制され、本剤の吸収が増加する可能性がある。 |
| アルコール | アルコールとの同時服用により本剤の徐放性が失われるおそれがあるため、本剤服用中は飲酒を避けるよう指導すること。 | in vitroの溶出試験において、アルコール存在下で徐放錠から成分が急速に溶出したとの報告がある。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 急性腎障害(頻度不明)
11.1.2 皮膚粘膜眼症候群(Stevens-Johnson症候群)(頻度不明)
11.1.3 薬剤性過敏症症候群(頻度不明)
初期症状として発疹、発熱がみられ、さらに肝機能障害等の臓器障害、リンパ節腫脹、白血球増加、好酸球増多、異型リンパ球出現等を伴う遅発性の重篤な過敏症状があらわれることがあるので、観察を十分に行い、このような症状があらわれた場合には投与を中止し、適切な処置を行うこと。なお、発疹、発熱、肝機能障害等の症状が再燃あるいは遷延化することがあるので注意すること。
11.1.4 肝炎、肝機能障害、黄疸(いずれも頻度不明)
11.1.5 横紋筋融解症(頻度不明)
筋肉痛、脱力感、CK上昇、血中及び尿中ミオグロビン上昇等があらわれた場合には、投与を中止し、適切な処置を行うこと。また、横紋筋融解症による急性腎障害の発症に注意すること。
11.1.6 アナフィラキシー(頻度不明)
アナフィラキシー(血管性浮腫、呼吸困難等)があらわれることがある。
11.2 その他の副作用
| 5%以上 | 1〜5%未満 | 1%未満 | |
| 血液及びリンパ系障害 | 好酸球数増加、血小板数増加 | ||
| 心臓障害 | 動悸 | ||
| 耳及び迷路障害 | 回転性めまい | ||
| 眼障害 | 霧視 | ||
| 胃腸障害 | 悪心、口内乾燥、下痢、便秘 | 鼓腸、消化不良、腹部不快感、嘔吐、上腹部痛、腹痛、胃食道逆流性疾患 | |
| 全身障害及び投与局所様態 | 疲労、易刺激性、体重増加 | 異常感、酩酊感、末梢性浮腫、倦怠感、無力症、体重減少 | |
| 肝胆道系障害 | ALT上昇、γ-GTP上昇、AST上昇 | ||
| 代謝及び栄養障害 | CK上昇 | 食欲亢進、尿酸上昇 | |
| 筋骨格系及び結合組織障害 | 四肢痛、筋肉痛、筋痙縮、関節痛、背部痛 | ||
| 神経系障害 | 傾眠(19.3%)、浮動性めまい(13.0%) | 頭痛、鎮静、平衡障害 | 注意力障害、錯感覚、振戦、嗜眠、味覚異常、構語障害、運動失調 |
| 精神障害 | 失見当識、うつ病、不眠症、不安、リビドー減退 | 異常な夢 | |
| 腎及び尿路障害 | BUN上昇 | ||
| 皮膚及び皮下組織障害 | 発疹、そう痒症 | ||
| 血管障害 | 高血圧 |
13. 過量投与
13.1 症状
外国において本剤を6gまで投与した例が報告されている。過量投与後にみられた主な症状は、精神運動制止遅滞、回転性めまい、鎮静及び傾眠である。
13.2 処置
本剤の活性代謝物であるガバペンチンは血液透析により除去可能であり、発現している症状の程度に応じて血液透析の実施を考慮すること。[16.6.2参照]
14. 適用上の注意
14.1 薬剤調製時の注意
PTP包装から取り出し無包装状態で高温・多湿下の条件に放置すると、品質の低下が認められるため、分包しないこと。
14.2 薬剤交付時の注意
14.2.1 PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
14.2.2 本剤は徐放性製剤であるため、割ったり、砕いたり、すりつぶしたりしないで、そのままかまずに服用するよう指導すること。割ったり、砕いたり、すりつぶしたりして服用すると、本剤の徐放性が失われるおそれがある。
15. その他の注意
15.1 臨床使用に基づく情報
15.1.1 海外で実施された本剤の活性代謝物であるガバペンチンを含む複数の抗てんかん薬における、てんかん、精神疾患等を対象とした199のプラセボ対照臨床試験の検討結果において、自殺念慮及び自殺企図の発現のリスクが、抗てんかん薬の服用群でプラセボ群と比較して約2倍高く(抗てんかん薬服用群:0.43%、プラセボ群:0.24%)、抗てんかん薬の服用群では、プラセボ群と比べ1,000人あたり1.9人多いと計算された(95%信頼区間:0.6-3.9)。また、てんかん患者のサブグループでは、プラセボ群と比べ1,000人あたり2.4人多いと計算されている注1)。
15.1.2 臨床試験において、本剤の依存性の可能性は評価されていない。
15.2 非臨床試験に基づく情報
15.2.1 非臨床薬物動態試験において、本薬はラットの眼球に投与後24時間以上にわたって分布したが、投与後72時間に眼球から消失することが確認された。また、マウス3カ月間、ラット6カ月間及びサル9カ月間反復投与毒性試験において眼球の変化は認められなかった。眼に関する副作用の発現率は、12週間投与の国内臨床試験ではプラセボ群3.4%に対し、本剤600mg/日群では認められず、900mg/日群で1.7%、1,200mg/日群で1.8%、長期投与では3.3%であり、12週間投与の海外臨床試験では、プラセボ群で認められなかったのに対し、本剤600mg/日群で0.6%、1,200mg/日群で4.1%、1,800mg/日群で2.6%、2,400mg/日群で8.9%、長期投与では1.4%であった注2)。[8.3参照]
15.2.2 ラットのがん原性試験(2年間強制経口投与)において発がん性が認められている。5,000mg/kg/day(本剤の1日臨床用量600mgにおけるヒト全身曝露量の90倍相当)の用量で膵臓腺房細胞腫瘍(腺腫あるいは腺癌)の発生が雌雄ともに増加し、その数は雌よりも雄に多かった。2,000mg/kg/day(本剤の1日臨床用量600mgにおけるヒト全身曝露量の40倍相当)の用量では雄においてこの膵臓腺房細胞腫瘍が増加していた。500mg/kg/day(本剤の1日臨床用量600mgにおけるヒト全身曝露量の10倍相当)では影響は認められなかった。マウスでは雌雄ともに発がん性は認められなかった。本剤の活性代謝物であるガバペンチンでも雄ラットに2,000mg/kg/day(本剤の1日臨床用量600mgにおけるヒト全身曝露量の40倍相当)を投与した際に同様の膵臓腺房細胞腫瘍の発生が報告されている。1,000mg/kg/day(本剤の1日臨床用量600mgにおけるヒト全身曝露量の30倍相当)ではこの腫瘍の増加は報告されていない。
注1)本剤の承認された効能又は効果は「中等度から高度の特発性レストレスレッグス症候群(下肢静止不能症候群)」である。
注2)本剤の承認された用法及び用量は「通常、成人にはガバペンチン エナカルビルとして1日1回600mgを夕食後に経口投与する」である。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
健康成人(各投与量6例)に本剤600、1,200あるいは1,800mgを注)空腹時に単回経口投与したとき、本剤の活性代謝物であるガバペンチンの全血中濃度は投与後4〜6時間で最高値に達し、消失半減期は4〜6時間であった。ガバペンチンのCmax及びAUCは用量の増加に伴って上昇した。なお、未変化体としては全血中にほとんど存在しなかった1)。
| 投与量 | n | Cmax(μg/mL) | Tmax(h) | t1/2(h) | AUCinf(μg・h/mL) |
| 600mg | 6 | 2.47±0.76 | 4.35±1.35 | 4.89±0.27 | 21.3±6.24 |
| 1,200mg | 6 | 5.08±1.26 | 5.67±0.82 | 5.31±0.88 | 47.1±6.12 |
| 1,800mg | 6 | 8.59±2.45 | 4.52±1.25 | 5.68±0.91 | 83.3±16.6 |
単回投与時の全血中ガバペンチン濃度推移
16.1.2 反復投与
健康成人10例に本剤1,200mgを注)1日1回、食後に5日間反復経口投与したときの最終投与時では、本剤の活性代謝物であるガバペンチンの全血中濃度は投与後5.2時間で最高値に達し、消失半減期は5.6時間であった2)(外国人データ)。
| Cmax(μg/mL) | Tmax(h) | t1/2(h) | AUCτ(μg・h/mL) |
| 6.10±1.29 | 5.20±1.14 | 5.64±1.08 | 63.9±11.7 |
16.1.3 レストレスレッグス症候群患者
レストレスレッグス症候群患者に本剤600、1,200、1,800あるいは2,400mgを注)1日1回、食後に反復経口投与したときの血漿中ガバペンチン濃度は、投与4週目及び12週目で明らかな変化は認められず、投与後6〜9時間の間に最高値を示し、消失半減期は5〜7時間であった。ガバペンチンのCmax及びAUCは用量にほぼ比例して上昇した3)(外国人データ)。
| 投与量 | n | Cmax(μg/mL) | Tmax(h) | t1/2(h) | AUC24h(μg・h/mL) |
| 600mg | 32 | 4.14±1.19 | 6.96±3.76 | 6.27±1.77 | 51.4±16.5 |
| 1,200mg | 30 | 7.15±2.76 | 8.72±3.68 | 6.63±2.23 | 95.7±38.5 |
| 1,800mg | 30 | 12.0±3.83 | 8.00±2.58 | 5.89±1.36 | 146±41.4 |
| 2,400mg | 31 | 13.3±3.83 | 8.13±3.20 | 6.09±1.28※ | 173±54.4※ |
16.2 吸収
16.2.1 吸収率
健康成人6例に本剤1,800mgを注)食後に単回経口投与したときのガバペンチンとしての平均累積尿中排泄率は73%であり、本剤経口投与時の吸収率は良好であると考えられた1)。
16.2.2 食事の影響
健康成人18例において、食後(高脂肪食)に本剤1,200mgを注)単回経口投与したときのCmax及びAUCは空腹時に比べ約40%上昇した4)。
| Cmax(μg/mL) | Tmax(h) | t1/2(h) | AUCinf(μg・h/mL) | |
| 空腹時投与 | 5.49±1.25 | 5.3±1.2 | 5.8±0.8 | 55.3±10.2 |
| 食後投与 | 7.55±0.92 | 6.1±1.7 | 5.1±0.4 | 76.2±6.7 |
16.3 分布
16.4 代謝
16.5 排泄
健康成人6例に本剤の14C-標識体600mgを食後に単回経口投与したとき、投与した総放射能の94.1%が尿中へ、5.2%が糞中へ排泄された。血液及び尿中総放射能の大部分は本剤の活性代謝物であるガバペンチンであり、本剤の主な排泄経路はガバペンチンとしての腎臓からの尿中排泄であると考えられた10)(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
腎機能の異なる被験者(12例)に本剤600mgを食後に単回経口投与したとき、腎機能の低下に伴って血漿中ガバペンチンの消失半減期が延長し、Cmax及びAUCが増加した。また、各被験者の経口クリアランス(CL/F)及び腎クリアランス(CLr)と腎機能の指標であるクレアチニンクリアランス(Ccr)の間には相関関係が認められ、Ccrの低下に伴ってCL/F及びCLrが低下した11)。[2.2、7.1、7.2、9.2.1-9.2.3参照]
| 腎機能分類(Ccr:mL/min) | 正常者※(Ccr≧90) | 軽度障害患者(90>Ccr≧60) | 中等度障害患者(60>Ccr≧30) | 高度障害患者※(30>Ccr≧15) |
| n | 1 | 4 | 6 | 1 |
| Cmax(μg/mL) | 4.35 | 4.94±1.37 | 6.46±1.48 | 8.70 |
| Tmax(h) | 8.0 | 7.3±1.5 | 10.5±4.4 | 8.1 |
| AUCinf(μg・h/mL) | 59.0 | 72.0±12.7 | 165.6±35.3 | 235.4 |
| t1/2(h) | 7.4 | 8.3±1.6 | 14.7±3.4 | 16.4 |
| CL/F(L/h) | 5.29 | 4.45±0.81 | 1.96±0.40 | 1.33 |
| Vz/F(L) | 56.2 | 54.4±19.8 | 40.6±8.8 | 31.5 |
| CLr(L/h) | 4.11 | 3.31±0.23 | 1.62±0.30 | 0.79 |
16.6.2 血液透析患者
16.7 薬物相互作用
16.7.1 ナプロキセン
健康成人10例を対象に、ナプロキセン(1回500mg、1日2回投与)と本剤(1回1,200mg、1日1回食後投与)を注)5日間反復経口投与したとき、本剤はナプロキセンの薬物動態に影響を及ぼさず、またナプロキセンも本剤投与時のガバペンチンの薬物動態に影響を及ぼさなかった2)(外国人データ)。
16.7.2 シメチジン
健康成人12例を対象に、シメチジン(1回400mg、1日4回投与)と本剤(1回1,200mg、1日1回食後投与)を注)反復経口投与したとき、本剤単独投与時と比較してガバペンチンのAUCτは24%増加したが、Cmaxは変化しなかった。また、本剤の投与はシメチジンの薬物動態に影響を及ぼさなかった12)(外国人データ)。
注)本剤の承認された用法及び用量は「通常、成人にはガバペンチン エナカルビルとして1日1回600mgを夕食後に経口投与する。」である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国内第II/III相試験
レストレスレッグス症候群患者469例を対象とした12週間のプラセボ対照無作為化二重盲検並行群間比較試験の結果、主要評価項目である最終観察時のIRLS合計スコアの変化量は、プラセボ群−8.96±7.286、本剤600mg群−11.10±7.921であり、プラセボ群との差とその95%信頼区間は−2.14[−4.097,−0.189]であった。副作用の発現率は、プラセボ群で50.9%(59/116例)、本剤600mg群で56.7%(68/120例)である。5%以上の副作用は、プラセボ群で傾眠16.4%(19/116例)、浮動性めまい6.9%(8/116例)及び頭痛5.2%(6/116例)、本剤600mg群で浮動性めまい25%(30/120例)、傾眠19.2%(23/120例)及び悪心5.0%(6/120例)であった13)。
17.1.2 海外第III相試験
レストレスレッグス症候群患者322例を対象とした12週間のプラセボ対照無作為化二重盲検並行群間比較試験の結果、主要評価項目である最終評価時のIRLS合計スコアの変化量はプラセボ群−9.8±7.69、本剤600mg群−13.8±8.09であり、プラセボ群と本剤600mg群の対比較において、統計学的な有意差が認められた(p<0.0001、施設及びベースライン値で調整した共分散分析)。また、最終評価時におけるICGIスケールでのレスポンダー率は、プラセボ群で44.8%(43/96例)、600mg群で72.8%(83/114例)、オッズ比とその95%信頼区間は3.322[1.841,5.992]であり、プラセボ群と本剤600mg群の対比較において、統計学的な有意差が認められた(p<0.0001、投与群及びプールした施設を説明変数としたロジスティック回帰モデル)。副作用の発現率は、プラセボ群で41.7%(40/96例)、本剤600mg群で54.8%(63/115例)である。5%以上の副作用は、プラセボ群なし、本剤600mg群で傾眠20.0%(23/115例)、浮動性めまい9.6%(11/115例)及び頭痛9.6%(11/115例)であった14)。
17.1.3 海外第III相長期投与試験
レストレスレッグス症候群患者を対象として本剤600〜1,800mg注)を52週間投与した結果、IRLS合計スコアの推移は下表のとおりであった。
| 評価時期 | 例数 | IRLS合計スコア | ベースラインからの変化量 |
| ベースラインa) | 573 | 23.2±5.03 | − |
| 0週時b) | 573 | 10.4±8.13 | −12.8±8.64 |
| 1週時 | 546 | 9.0±7.50 | −14.2±8.19 |
| 4週時 | 526 | 7.5±7.24 | −15.7±7.77 |
| 12週時 | 472 | 7.1±7.23 | −16.1±8.14 |
| 24週時 | 444 | 6.9±7.14 | −16.4±7.71 |
| 52週時 | 379 | 6.5±7.40 | −16.8±8.21 |
| 最終評価時(LOCF) | 573 | 8.0±8.29 | −15.2±8.85 |
副作用の発現率は、先行試験での本剤未投与群で66.5%(131/197例)、本剤投与群で49.2%(185/376例)である。5%以上の副作用は、本剤未投与群で傾眠26.9%(53/197例)、浮動性めまい19.8%(39/197例)、疲労6.6%(13/197例)、下肢静止不能症候群5.6%(11/197例)及び頭痛5.1%(10/197例)、本剤投与群で傾眠15.7%(59/376例)及び浮動性めまい6.1%(23/376例)であった15)。
17.2 製造販売後調査等
17.2.1 国内製造販売後臨床試験
レストレスレッグス症候群患者375例を対象とした12週間のプラセボ対照無作為化二重盲検並行群間比較試験の結果、主要評価項目である治療期最終時点におけるIRLS合計スコアの変化量の調整済み平均値とその95%信頼区間は、本剤600mg群で−11.7[−12.6,−10.7]、プラセボ群で−10.5[−11.4,−9.5]であった。その差とその95%信頼区間は−1.2[−2.6,0.2]であり、統計的に有意な差は認められなかった(MMRM解析、p=0.088、有意水準両側0.05)。副作用の発現率は、プラセボ群で19.4%(36/186例)、本剤600mg群で31.7%(60/189例)である。5%以上の副作用は、プラセボ群で傾眠7.0%(13/186例)、本剤600mg群で傾眠13.2%(25/189例)及び浮動性めまい10.1%(19/189例)であった16)。[5.2参照]
注)本剤の承認された用法及び用量は「通常、成人にはガバペンチン エナカルビルとして1日1回600mgを夕食後に経口投与する。」である。
18. 薬効薬理
18.1 作用機序
19. 有効成分に関する理化学的知見
19.1. ガバペンチン エナカルビル
| 一般的名称 | ガバペンチン エナカルビル |
|---|---|
| 一般的名称(欧名) | Gabapentin Enacarbil |
| 化学名 | (1-{[({(1RS)-1-[(2-Methylpropanoyl)oxy]ethoxy}carbonyl)amino]methyl}cyclohexyl)acetic acid |
| 分子式 | C16H27NO6 |
| 分子量 | 329.39 |
| 融点 | 約67℃ |
| 物理化学的性状 | ガバペンチン エナカルビルは白色の結晶又は粉末である。アセトニトリル、N,N-ジメチルホルムアミド、メタノール又はエタノール(99.5)に極めて溶けやすく、水に極めて溶けにくい。また、吸湿性を認めない。 |
| KEGG DRUG |
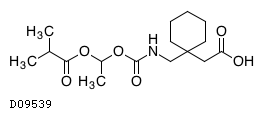
|
20. 取扱い上の注意
20.1 本品は熱により黄変することがあるので、高温での保存を避け、涼しい場所で保存すること。
20.2 本品はアルミ袋と乾燥剤により品質保持をはかっているので、内袋開封後は乾燥剤が封入された専用の保管袋で高温・湿気を避けて保存し、なるべく速やかに使用すること。
22. 包装
56錠(14錠×4、乾燥剤入り)
23. 主要文献
- 社内報告書:健康成人・単回投与試験(2012年1月18日承認 CTD2.7.6.3)
- 社内報告書:健康成人・薬物相互作用検討試験(2012年1月18日承認 CTD2.7.6.10)
- 社内報告書:レストレスレッグス症候群患者(外国人)・第II相二重盲検比較試験(2012年1月18日承認 CTD2.7.6.17)
- 社内報告書:健康成人・食事の影響検討試験(2012年1月18日承認 CTD2.7.6.1)
- 社内報告書:健康成人・反復投与試験(2012年1月18日承認 CTD2.7.6.4)
- Radulovic,L.L.et al., Drug Metab.Dispos., 23 (4), 441-448, (1995) »PubMed
- Cundy,C.K.et al., J.Pharmacol.Exp.Ther., 311 (1), 315-323, (2004) »PubMed
- 社内報告書:In vitro代謝及び蛋白結合率検討試験(2012年1月18日承認 CTD2.7.2.2.1.2、CTD2.7.2.2.1.3)
- 社内報告書:In vitro酵素誘導検討試験(2012年1月18日承認 CTD2.7.2.2.1.5)
- 社内報告書:健康成人・マスバランス試験(2012年1月18日承認 CTD2.7.6.5)
- 社内報告書:腎機能障害患者・薬物動態検討試験(2012年1月18日承認 CTD2.7.6.8)
- 社内報告書:健康成人・薬物相互作用検討試験(2012年1月18日承認 CTD2.7.6.11)
- 社内報告書:レストレスレッグス症候群患者・第II/III相二重盲検比較試験(2012年1月18日承認 CTD2.7.6.14)
- 社内報告書:レストレスレッグス症候群患者(外国人)・第III相二重盲検比較試験(2012年1月18日承認 CTD2.7.6.19)
- 社内報告書:レストレスレッグス症候群患者(外国人)・長期投与試験(2012年1月18日承認 CTD2.7.6.22)
- 社内報告書:レストレスレッグス症候群患者・製造販売後臨床試験
- Marais,E.et al., Mol.Pharmacol., 59 (5), 1243-1248, (2001) »PubMed
- Fink,K.et al., Br.J.Pharmacol., 130 (4), 900-906, (2000) »PubMed »DOI
24. 文献請求先及び問い合わせ先
文献請求先
アステラス製薬株式会社
メディカルインフォメーションセンター
〒103-8411
東京都中央区日本橋本町2丁目5番1号
電話:フリーダイヤル 0120-189-371
製品情報問い合わせ先
アステラス製薬株式会社
メディカルインフォメーションセンター
〒103-8411
東京都中央区日本橋本町2丁目5番1号
電話:フリーダイヤル 0120-189-371
26. 製造販売業者等
26.1 製造販売
アステラス製薬株式会社
東京都中央区日本橋本町2丁目5番1号