医薬品情報
| 総称名 | ヴォトリエント |
|---|---|
| 一般名 | パゾパニブ塩酸塩 |
| 欧文一般名 | Pazopanib Hydrochloride |
| 製剤名 | パゾパニブ塩酸塩錠 |
| 薬効分類名 | 抗悪性腫瘍剤 キナーゼ阻害剤 |
| 薬効分類番号 | 4291 |
| ATCコード | L01EX03 |
| KEGG DRUG |
D05380
パゾパニブ塩酸塩
|
| JAPIC | 添付文書(PDF) |
添付文書情報2024年2月 改訂(第2版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ヴォトリエント錠200mg | Votrient Tablets 200mg | ノバルティスファーマ | 4291028F1023 | 3331.9円/錠 | 劇薬, 処方箋医薬品 |
1. 警告
2. 禁忌
次の患者には投与しないこと
2.1 本剤の成分に対し過敏症の既往歴のある患者
2.2 妊婦又は妊娠している可能性のある女性[9.5参照]
4. 効能または効果
5. 効能または効果に関連する注意
<悪性軟部腫瘍>
5.1 本剤の化学療法未治療例における有効性及び安全性は確立していない。
<根治切除不能又は転移性の腎細胞癌>
5.3 本剤の術後補助化学療法における有効性及び安全性は確立していない。
6. 用法及び用量
通常、成人にはパゾパニブとして1日1回800mgを食事の1時間以上前又は食後2時間以降に経口投与する。なお、患者の状態により適宜減量する。
7. 用法及び用量に関連する注意
7.1 他の抗悪性腫瘍剤(サイトカイン製剤を含む)との併用について、有効性及び安全性は確立していない。
7.2 食後に本剤を投与した場合、Cmax及びAUCが上昇するとの報告がある。食事の影響を避けるため、用法及び用量を遵守して服用すること。[16.2.1参照]
7.3 副作用の発現により用量を減量して投与を継続する場合は、症状、重症度等に応じて、200mgずつ減量すること。また、本剤を減量後に増量する場合は、200mgずつ増量すること。ただし、800mgを超えないこと。
7.4 臨床試験において、中等度の肝機能障害を有する患者に対する最大耐用量は200mgであることが確認されており、中等度以上の肝機能障害を有する患者に対して本剤200mgを超える用量の投与は、最大耐用量を超えるため推奨されない。[1.3、9.3.1、16.6.2、17.3.1参照]
7.5 本剤を服用中に肝機能検査値異常が発現した場合は、以下の基準を考慮して、休薬、減量又は中止すること。[8.1、11.1.1参照]
| 肝機能検査値 | 処置 |
| 3.0×ULN≦ALT≦8.0×ULN | 投与継続(Grade 1以下あるいは投与前値に回復するまで1週間毎に肝機能検査を実施) |
| ALT>8.0×ULN | Grade 1以下あるいは投与前値に回復するまで投与を中断し、投与を再開する場合は、400mgの投与とする。 再開後、肝機能検査値異常(ALT>3.0×ULN)が再発した場合は、投与を中止する。 |
| ALT>3.0×ULN、かつ総ビリルビン>2.0×ULN (直接ビリルビン>35%) | 投与中止(Grade 1以下あるいは投与前値に回復するまで経過を観察) |
8. 重要な基本的注意
8.2 高血圧があらわれることがあるので、本剤の投与開始前及び投与期間中は定期的に血圧測定を行い、血圧を十分観察すること。また、高血圧クリーゼがあらわれることがあるので、血圧の推移等に十分注意して投与すること。[9.1.1、11.1.2参照]
8.4 QT間隔延長、心室性不整脈(Torsade de pointesを含む)があらわれることがあるので、本剤の投与開始前及び投与中は定期的に心電図検査及び電解質測定を行い、必要に応じて、電解質(カルシウム、マグネシウム、カリウム)を補正すること。[9.1.3、11.1.4参照]
8.5 創傷治癒を遅らせる可能性があるので、外科的処置が予定されている場合には、外科的処置の前に本剤の投与を中断すること。外科的処置後の投与再開は、患者の状態に応じて判断すること。[9.1.7、11.1.13参照]
8.6 甲状腺機能障害があらわれることがあるので、本剤の投与開始前及び投与期間中は定期的に甲状腺機能の検査を実施すること。[11.1.10参照]
8.7 ネフローゼ症候群、蛋白尿があらわれることがあるので、本剤の投与開始前及び投与期間中は定期的に尿蛋白を観察すること。[11.1.11参照]
8.8 毛髪変色又は皮膚の色素脱失があらわれることがあるので、本剤を投与する場合にはその内容を適切に患者に説明すること。また、剥脱性皮膚炎、手足症候群等の皮膚障害があらわれることがあるので、十分に観察を行い、異常が認められた場合には適切な処置を行うこと。必要に応じて患者に皮膚科受診等を指導すること。
8.9 血栓性微小血管症があらわれることがあるので、定期的に検査を行うなど観察を十分に行うこと。[11.1.15参照]
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 高血圧の患者
9.1.2 心機能障害のリスク因子を有する患者(特に、アントラサイクリン系薬剤等の心毒性を有する薬剤、及び放射線治療による治療歴のある患者)
9.1.3 QT間隔延長の既往のある患者
9.1.4 血栓塞栓症又はその既往歴のある患者
9.1.5 脳転移を有する患者
臨床試験において、転移部位からの出血が報告されている。[11.1.8参照]
9.1.6 肺転移を有する患者
気胸が悪化又は発現するおそれがある。また、臨床試験において、転移部位からの出血が報告されている。[11.1.8参照]
9.1.7 外科的処置後、創傷が治癒していない患者
9.2 腎機能障害患者
9.2.1 重度の腎機能障害患者
臨床試験では除外されている。
9.3 肝機能障害患者
9.4 生殖能を有する者
妊娠可能な女性に対しては、本剤投与中及び投与終了後一定期間は避妊を行うよう指導すること。[9.5参照]
9.5 妊婦
9.6 授乳婦
授乳しないことが望ましい。ヒトで乳汁移行に関するデータはないが、BCRPの基質であるため、乳汁移行の可能性がある。
9.7 小児等
小児等を対象とした臨床試験は実施していない。本剤の作用機序より、出生後早期の発達において臓器の成長や成熟に重大な影響を与えるおそれがある。
9.8 高齢者
患者の状態を観察しながら慎重に投与すること。一般に生理機能が低下していることが多い。
10. 相互作用
相互作用序文
薬物代謝酵素用語
CYP3A4
薬物代謝酵素用語
CYP1A2
薬物代謝酵素用語
CYP2C8
薬物代謝酵素用語
CYP2B6
薬物代謝酵素用語
CYP2E1
薬物代謝酵素用語
UGT1A1
薬物代謝酵素用語
OATP1B1
薬物代謝酵素用語
P-糖蛋白質(Pgp)
薬物代謝酵素用語
BCRP
10.2 併用注意
| プロトンポンプ阻害剤 エソメプラゾール等 | エソメプラゾールとの併用により、本剤のAUC及びCmaxがそれぞれ約40%及び42%低下したとの報告があるので、プロトンポンプ阻害剤との併用は可能な限り避けること。 | プロトンポンプ阻害剤が胃内の酸分泌を抑制することで、本剤の溶解度が低下し吸収が低下する可能性がある。 |
| CYP3A4阻害剤 ケトコナゾール等 | ケトコナゾールとの併用により、本剤のAUC及びCmaxは、それぞれ約66%及び45%増加した。 CYP3A4阻害作用のない又は弱い薬剤への代替を考慮すること。併用が避けられない場合には、副作用の発現・増強に注意し、減量等を考慮すること。 | これらの薬剤がCYP3A4活性を阻害することにより、本剤の代謝が阻害され、血中濃度が上昇する可能性がある。 |
| CYP3A4阻害剤 グレープフルーツ(ジュース) | 本剤投与時はグレープフルーツ(ジュース)を摂取しないよう注意すること。 | これらの薬剤がCYP3A4活性を阻害することにより、本剤の代謝が阻害され、血中濃度が上昇する可能性がある。 |
| CYP3A4誘導剤 カルバマゼピン フェニトイン等 | カルバマゼピン、フェニトイン等との併用により、本剤のAUC及びCmaxは、それぞれ約54%及び35%低下した。 CYP3A4誘導作用のない又は弱い薬剤への代替を考慮すること。併用に際しては、本剤の有効性が減弱する可能性があることを考慮すること。 | これらの薬剤がCYP3A4活性を誘導することにより、本剤の代謝が誘導され、血中濃度が低下する可能性がある。 |
| パクリタキセル | 本剤は血漿中パクリタキセルのAUC及びCmaxをそれぞれ約26%及び31%増加させた。 | 本剤がCYP3A4及びCYP2C8活性を阻害することにより、パクリタキセルの代謝が阻害され、血中濃度が上昇する可能性がある。 |
| ラパチニブ | ラパチニブとの併用により本剤のAUC及びCmaxは、それぞれ約59%及び51%増加した。 | ラパチニブはCYP3A4、Pgp及びBCRPの基質であり阻害作用を有することによる。 |
| シンバスタチン | 併用によりALTが上昇するおそれがある。 | 機序は不明である。 |
| 抗不整脈薬 キニジン プロカインアミド ジソピラミド等 QT間隔を延長させる可能性のある薬剤 クラリスロマイシン イミプラミン ピモジド等 | QT間隔延長や心室性不整脈をおこすおそれがある。 | 本剤及びこれらの薬剤はいずれもQT間隔を延長させるおそれがあり、併用により作用が増強する可能性がある。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 肝不全(頻度不明)、肝機能障害(28.4%)
11.1.2 高血圧(42.0%)、高血圧クリーゼ(0.6%)
11.1.3 心機能障害(2.8%)
11.1.5 動脈血栓性事象(1.8%)
心筋梗塞、狭心症、虚血性脳卒中、一過性脳虚血発作、心筋虚血等の動脈血栓性事象があらわれることがある。[9.1.4参照]
11.1.6 静脈血栓性事象(1.1%)
静脈血栓症及び肺塞栓症があらわれることがある。[9.1.4参照]
11.1.7 動脈解離(頻度不明)
大動脈解離を含む動脈解離があらわれることがある1)。
11.1.8 出血(13.2%)
11.1.9 消化管穿孔(頻度不明)、消化管瘻(0.5%)
11.1.10 甲状腺機能障害(12.6%)[8.6参照]
11.1.11 ネフローゼ症候群(0.1%)、蛋白尿(12.5%)[8.7参照]
11.1.12 感染症(8.6%)
好中球減少の有無にかかわらず重篤な感染症があらわれることがある。
11.1.13 創傷治癒遅延(0.4%)
11.1.14 間質性肺炎(0.1%)
11.1.15 血栓性微小血管症(0.1%)
血栓性血小板減少性紫斑病、溶血性尿毒症症候群等の血栓性微小血管症があらわれることがある。破砕赤血球を伴う貧血、血小板減少、腎機能障害等が認められた場合には、投与を中止し、適切な処置を行うこと。[8.9参照]
11.1.16 可逆性後白質脳症症候群(頻度不明)
可逆性後白質脳症症候群に一致する徴候や症状(高血圧(伴わない例もある)、頭痛、覚醒低下、精神機能変化、及び皮質盲を含めた視力消失など)が認められた場合は、本剤の投与を中止し、高血圧管理を含め、適切な処置を行うこと。
11.1.17 膵炎(3.8%)
膵炎を示唆する症状があらわれた場合には、投与を中止し、適切な処置を行うこと。
11.1.18 網膜剥離(0.1%)
飛蚊症、光視症、視野欠損、視力低下等が認められた場合には、眼科検査を実施し、投与を中止するなど適切な処置を行うこと。
注)副作用の頻度については、悪性軟部腫瘍患者を対象とした国際共同第III相臨床試験並びに腎細胞癌患者を対象とした国際共同第III相臨床試験及び外国第III相臨床試験に基づき記載した。
11.2 その他の副作用
| 30%以上 | 5〜30%未満 | 5%未満 | |
| 代謝 | 食欲減退 | 体重減少 | 高カリウム血症、高血糖 |
| 神経系 | − | 味覚異常、頭痛 | 浮動性めまい、末梢性ニューロパチー、不眠症、傾眠 |
| 循環器 | − | − | 徐脈(無症候性) |
| 呼吸器 | − | 発声障害 | 呼吸困難、咳嗽、気胸 |
| 消化器 | 下痢、悪心 | 嘔吐、腹痛、消化不良、口内炎、便秘 | 口内乾燥、腹部膨満、口腔咽頭痛、胃炎、しゃっくり、痔核、嚥下障害、鼓腸 |
| 皮膚 | 毛髪変色 | 手掌・足底発赤知覚不全症候群、発疹、脱毛症、皮膚色素減少、皮膚乾燥 | 剥脱性発疹、そう痒症、皮膚障害、爪の障害、ざ瘡、皮膚潰瘍、毛髪成長異常 |
| 筋骨格 | − | 筋骨格痛 | 筋肉痛、関節痛、筋痙縮 |
| 血液 | − | 血小板減少症、好中球減少症、白血球減少症、貧血 | リンパ球減少症、赤血球増加症 |
| 臨床検査 | − | − | 血中クレアチニン増加、リパーゼ増加、血中アルカリホスファターゼ増加、LDH異常、血中ナトリウム減少、血中カルシウム減少、血中マグネシウム減少、血中尿素増加、血中リン減少、血中ブドウ糖減少、血中アルブミン減少 |
| その他 | 疲労 | 粘膜炎、無力症 | 末梢性浮腫、顔面浮腫、胸痛、霧視、ほてり、発熱、多汗症、脱水、腫瘍疼痛、浮腫、悪寒、挫傷、不規則月経 |
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
15. その他の注意
15.1 臨床使用に基づく情報
本剤とペメトレキセド及びラパチニブを併用した固形癌患者を対象とした臨床試験において、毒性の増大、死亡率の増加が懸念されたため早期に中止されている。
15.2 非臨床試験に基づく情報
15.2.1 マウスを用いた反復投与毒性試験において、1000mg/kg/日の雌で増殖性変化(好酸性変異肝細胞巣及び肝細胞腺腫がそれぞれ2及び1例)が認められた。
15.2.2 幼若ラットの生後9〜21日まで投与した試験において、30mg/kg/日以上で体重増加抑制及び早期死亡が認められ、生後21〜62日まで投与した試験においては、10mg/kg/日以上で大腿骨の短小が認められた(これらの用量はいずれも成熟ラットにおいて同様の影響がみられた用量よりも低用量)。
16. 薬物動態
16.1 血中濃度
16.1.1 経口投与
固形癌患者13例に本剤を1日1回反復経口投与したときの薬物動態パラメータは下表のとおりであった。本剤の投与量と1日目のCmax及びAUC0-24との間に、用量比例性は認められなかった。また、本剤の血漿中からの消失は緩やかであり、反復投与で蓄積性を示すと考えられた2)。
| パラメータ | 400/800mg注1,注2) (N=3) | 800mg (N=7) | 1000mg注2) (N=3) | |||
| 1日目 | 22日目 | 1日目 | 22日目 | 1日目 | 22日目 | |
| Cmax(μg/mL) | 25.1 (34.0%) | 55.8 (35.2%) | 22.9 (69.5%) | 40.6 (47.7%) | 21.3 (118.1%) | 53.9 (55.4%) |
| tmax(hr) | 4.00 (3.00-23.72) | 2.50 (2.00-3.00) | 2.98 (1.97-5.95) | 2.52 (1.92-3.97) | 3.00 (2.97-3.00) | 4.00 (3.00-4.05) |
| AUC0-24(μg・hr/mL) | 402 (17.7%) | 962 (46.3%) | 325 (76.7%) | 677 (45.5%) | 305 (128.6%) | 759 (63.8%) |
| t1/2(hr) | 28.4 (35.9%) | 40.1 (67.2%) | 42.5 (31.6%) | 37.8 (47.2%) | 33.0 (23.8%) | 21.4 (60.7%) |
| C24(μg/mL) | 14.8 (12.7%) | 34.6 (47.2%) | 9.1 (90.1%) | 22.0 (48.4%) | 8.5 (139.6%) | 21.1 (80.5%) |
日本人固形癌患者に本剤800mgを単回及び反復経口投与したときの血漿中パゾパニブ濃度推移(平均値+標準偏差、n=7)
16.1.2 静脈内投与
外国人固形癌患者3例に承認用法・用量(1日1回経口800mg)とは異なる5mgを静脈内投与後のクリアランスは0.206〜0.347L/hr、Vssは9.2〜13.1Lであった3)。
16.2 吸収
16.2.1 食事の影響
16.3 分布
16.4 代謝
16.5 排泄
外国人固形癌患者3例に14C-標識体を単回経口投与したときの放射能の主排泄経路は糞中であり、投与後168時間までの排泄率は糞で約82.2%、尿で約2.6%であった。糞中の主成分は未変化体であり、投与量の平均67%であった3)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
腎機能低下者における本剤の特別な薬物動態は検討していない。クレアチニンクリアランス30〜150mL/minの外国人健康被験者及び癌患者408例で母集団薬物動態を解析した結果、腎機能による本剤のクリアランスに有意な影響はみられていない10)。
16.6.2 肝機能障害患者
外国人肝機能障害患者98例(肝機能正常者23例、軽度肝機能低下者23例、中等度肝機能低下者20例、重度肝機能低下者32例)に本剤を1日1回反復経口投与したときの安全性及び薬物動態について評価した(薬物動態解析対象例69例)。軽度の肝機能低下患者12例に本剤800mgを1日1回反復経口投与したときのCmax及びAUC0-24は肝機能正常者18例と同程度であった。肝機能低下患者(中等度11例及び重度14例)に200mgを1日1回反復経口投与したときのCmax及びAUC0-24の中央値は、肝機能正常者に800mgを1日1回反復経口投与したときのCmax及びAUC0-24のそれぞれ約43%及び29%、約18%及び15%であった11)。[1.3、7.4、9.3.1、17.3.1参照]
| 群 (N) | 投与量(mg) | Cmax(μg/mL) | tmax(hr) | C24(μg/mL) | AUC0-24(μg・hr/mL) |
| A (18) | 800 | 52.0 (17.1-85.7) | 2.8 (1.0-24.2) | 29.8 (10.3-750) | 888.2 (345.5-1482) |
| B (12) | 800 | 33.5 (11.3-104.2) | 3.0 (0.5-24.4) | 24.0 (8.3-74.6) | 774.2 (214.7-2034.4) |
| C (11) | 200 | 22.2 (4.2-32.9) | 2.0 (0.0-4.0) | 16.2 (3.1-24.2) | 256.8 (65.7-487.7) |
| D (14) | 200 | 9.4 (2.4-24.3) | 3.0 (1.0-8.0) | 5.7 (1.5-18.4) | 130.6 (46.9-473.2) |
16.7 薬物相互作用
In vitro試験で、本剤はCYP(1A2、2B6、2C8、2C9、2C19、2D6、2E1及び3A4)及びUGT1A1を阻害し、IC50はそれぞれ7.9〜18μM(3.5〜7.9μg/mL)及び1.2μM(0.5μg/mL)であった12)。
外国人固形癌患者24例を対象に本剤がCYP代謝に及ぼす影響について、各種CYPプローブを用いて検討した結果、本剤800mgの1日1回投与ではカフェイン(CYP1A2の基質)、ワルファリン(CYP2C9の基質)及びオメプラゾール(CYP2C19の基質)の薬物動態に意義のある影響を及ぼさなかった。一方、本剤はミダゾラム(CYP3A4の基質)のAUC及びCmaxを約30%増加させ、デキストロメトルファン(CYP2D6の基質)を経口投与後の尿中のデキストロメトルファン/デキストルファン比を33〜64%増加させた13)。
また、in vitro試験で、本剤はOATP1B1を阻害し、IC50は0.79μM(0.3μg/mL)であった14)。[10.参照]
外国人固形癌患者24例を対象に本剤がCYP代謝に及ぼす影響について、各種CYPプローブを用いて検討した結果、本剤800mgの1日1回投与ではカフェイン(CYP1A2の基質)、ワルファリン(CYP2C9の基質)及びオメプラゾール(CYP2C19の基質)の薬物動態に意義のある影響を及ぼさなかった。一方、本剤はミダゾラム(CYP3A4の基質)のAUC及びCmaxを約30%増加させ、デキストロメトルファン(CYP2D6の基質)を経口投与後の尿中のデキストロメトルファン/デキストルファン比を33〜64%増加させた13)。
また、in vitro試験で、本剤はOATP1B1を阻害し、IC50は0.79μM(0.3μg/mL)であった14)。[10.参照]
17. 臨床成績
17.1 有効性及び安全性に関する試験
<悪性軟部腫瘍>
17.1.1 国際共同第III相臨床試験
アントラサイクリン系薬剤を含む前治療に対して病勢進行が認められた転移病変を有する悪性軟部腫瘍患者を対象とした、二重盲検、プラセボ対照試験で、本剤800mgを1日1回経口投与した時の有効性及び安全性を評価した。本試験では、脂肪肉腫、横紋筋肉腫(多形型又は胞巣型を除く)、軟骨肉腫、骨肉腫、ユーイング腫瘍/未熟神経外胚葉性腫瘍、消化管間質腫瘍、隆起性皮膚線維肉腫、炎症性筋線維芽細胞肉腫、悪性中皮腫、子宮の中胚葉性混合腫瘍を組入れ対象から除外した。有効性解析対象例は369例(本剤群246例、プラセボ群123例)であり、このうち47例(本剤群31例、プラセボ群16例)が日本人であった。
主要評価項目である無増悪生存期間(PFS)の中央値は、本剤群で20.0週、プラセボ群で7.0週であり、本剤群のPFSはプラセボ群と比して有意に延長した(ハザード比:0.35、95%信頼区間:0.26〜0.48、p<0.001、両側層別ログランク検定)。
PFSのKaplan-Meier曲線(全体集団、独立判定)
副次的評価項目である全生存期間(OS)の中央値は、本剤群で12.6ヵ月、プラセボ群で10.7ヵ月であり、事前に規定した有意水準(p≦0.04434)には至らなかった(ハザード比:0.87、95.57%信頼区間:0.67〜1.13、p=0.256、両側層別ログランク検定)15)。
副作用は本剤を投与された240例中(日本人31例を含む)219例(91.3%)に認められた。主な副作用は下痢(130例、54.2%)、疲労(126例、52.5%)、悪心(116例、48.3%)、高血圧(94例、39.2%)、毛髪変色(93例、38.8%)、食欲減退(82例、34.2%)、体重減少(73例、30.4%)であった。[5.2参照]
17.1.2 外国第II相臨床試験
再発又は難治性の悪性軟部腫瘍患者を対象とした、非盲検、非対照試験で、本剤800mg(製造販売用製剤とは異なる製剤)を1日1回経口投与した時の有効性及び安全性について、腫瘍組織型別(脂肪肉腫、平滑筋肉腫、滑膜肉腫、その他)に評価した。本試験では、胎児型横紋筋肉腫、軟骨肉腫、骨肉腫、ユーイング腫瘍/未熟神経外胚葉性腫瘍、消化管間質腫瘍、隆起性皮膚線維肉腫、炎症性筋線維芽細胞肉腫、悪性中皮腫、子宮の中胚葉性混合腫瘍を組入れ対象から除外した。有効性解析対象例は138例であった。
主要評価項目である投与12週時のprogression-free rate(PF率:その時点の評価がCR、PR又はSDであった被験者の割合)は以下のとおりであった16)17)。
| 脂肪肉腫 (N=19) | 平滑筋肉腫 (N=41) | 滑膜肉腫 (N=37) | その他 (N=41) | |
| 12週時評価 | ||||
| CR | 0 | 0 | 0 | 0 |
| PR | 0 | 1 | 4 | 1 |
| SD | 5 | 16 | 14 | 16 |
| PF率(%) (90%信頼区間) | 26 (11.0,47.6) | 41 (28.4,55.5) | 49 (34.3,63.2) | 41 (28.4,55.5) |
副作用は本剤を投与された142例中127例(89%)に認められた。主な副作用は高血圧(58例、41%)、皮膚色素減少(毛髪を含む)(53例、37%)、疲労(52例、37%)、悪心(51例、36%)、下痢(44例、31%)、嘔吐(35例、25%)であった。[5.2参照]
<根治切除不能又は転移性の腎細胞癌>
17.1.3 国際共同第III相臨床試験
全身治療による治療歴のない局所進行性又は転移性腎細胞癌患者に本剤800mgを1日1回経口投与した時の有効性及び安全性について、スニチニブ(50mgを1日1回4週間経口投与後に2週間休薬の6週間を1サイクルとして投与)を対照とした非盲検試験で評価した。有効性解析対象例は1110例注1)(本剤群557例、スニチニブ群553例)であり、このうち60例(本剤群29例、スニチニブ群31例)が日本人であった。組織学的分類では有効性解析対象1110例のうち1031例(92.9%)が淡明細胞型であった。
主要評価項目であるPFSの中央値は、本剤群で8.4ヵ月、スニチニブ群で9.5ヵ月であり、事前に規定した本剤群のスニチニブ群に対する非劣性の判断基準(ハザード比の95%信頼区間の上限値が1.25を下回る)を満たした(ハザード比:1.0466、95%信頼区間:0.8982〜1.2195)。
PFSのKaplan-Meier曲線(全体集団、独立判定)
副次的評価項目であるOSの中間解析(2012年5月時点)では、OSの中央値は、本剤群で28.4ヵ月、スニチニブ群で29.3ヵ月であった(ハザード比:0.908、95%信頼区間:0.762〜1.082、p=0.275、両側層別ログランク検定)18)。
副作用は本剤を投与された554例中(日本人29例を含む)538例(97%)に認められた。主な副作用は下痢(321例、58%)、疲労(273例、49%)、高血圧(240例、43%)、悪心(223例、40%)、手掌・足底発赤知覚不全症候群(159例、29%)であった。
注1)本試験に準じてアジア(中国、韓国及び台湾)で実施された第II相臨床試験の被験者183例(本剤群93例、スニチニブ群90例)を含む。
17.1.4 外国第III相臨床試験
局所進行性又は転移性腎細胞癌患者(未治療の患者又は1レジメンのサイトカイン治療歴のある患者)を対象とした、プラセボ対照、二重盲検試験で、本剤800mgを1日1回経口投与した時の有効性及び安全性を評価した。有効性解析対象例は435例(本剤群290例、プラセボ群145例)であり、組織学的分類ではこのうち434例(99.8%)が淡明細胞型であった。
主要評価項目であるPFSの中央値は、本剤群で9.2ヵ月、プラセボ群で4.2ヵ月であり、本剤群のPFSはプラセボ群と比して有意に延長した(ハザード比:0.46、95%信頼区間:0.34〜0.62、p<0.0000001、片側層別ログランク検定)。
PFSのKaplan-Meier曲線(全体集団、独立判定)
副次的評価項目であるOSの中央値は、本剤群で22.9ヵ月、プラセボ群で20.5ヵ月であり、有意な差は認められなかった(ハザード比:0.91、95%信頼区間:0.71〜1.16、p=0.224、片側層別ログランク検定)19)。
副作用は本剤を投与された290例中257例(89%)に認められた。主な副作用は下痢(44%)、高血圧(37%)、毛髪変色(37%)であった。
17.3 その他
17.3.1 肝機能障害患者を対象とした試験
外国人肝機能障害患者98例(肝機能正常者23例、軽度肝機能低下者23例、中等度肝機能低下者20例、重度肝機能低下者32例)に本剤を1日1回反復経口投与したときの安全性及び薬物動態について評価した(安全性解析対象例97例)。400mgコホートの中等度肝機能障害患者2/4例で用量制限毒性(Grade 4のAST増加1例、Grade 4のAST増加、Grade 4のALT増加及びGrade 3の高ビリルビン血症が1例)が認められ、中等度以上の肝機能障害患者での最大耐用量は200mgであった11)。[1.3、7.4、9.3.1、16.6.2参照]
| 投与量(mg) | 肝機能 | |||
| 正常 | 軽度低下 | 中等度低下 | 重度低下 | |
| 100 | − | − | − | 1/6例 ・ビリルビン増加(Grade 4) |
| 200 | − | − | 1/12例 ・ビリルビン増加(Grade 3) | 1/11例 ・下痢(Grade 3) |
| 400 | − | 1/6例 ・AST増加(Grade 4) | 2/4例 ・AST増加(Grade 4) ・AST増加(Grade 4)、ALT増加(Grade 4)、ビリルビン増加(Grade 3) | − |
| 800 | 0/18例 | 1/13例 ・胃出血(Grade 5) | − | − |
18. 薬効薬理
18.1 作用機序
18.1.1 パゾパニブは、血管内皮細胞増殖因子受容体(VEGFR-1、VEGFR-2及びVEGFR-3)、血小板由来増殖因子受容体(PDGFR-α及びPDGFR-β)、並びに幹細胞因子受容体(c-Kit)のリン酸化を阻害した。また、パゾパニブは、マウスに投与した時に肺のVEGFR-2のリン酸化を阻害した20)。
18.1.2 パゾパニブは、マウス脈絡膜にレーザー照射で誘発した血管新生、並びにウサギ脈絡膜に血管内皮細胞増殖因子及び塩基性線維芽細胞増殖因子で誘発した血管新生に対して、阻害作用を示した20)。
18.2 抗腫瘍効果
19. 有効成分に関する理化学的知見
19.1. パゾパニブ塩酸塩
| 一般的名称 | パゾパニブ塩酸塩 |
|---|---|
| 一般的名称(欧名) | Pazopanib Hydrochloride |
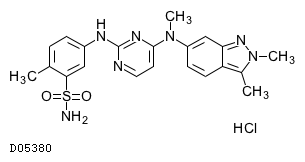
| 化学名 | 5-({4-[(2,3-ジメチル-2H-インダゾール-6-イル)(メチル)アミノ]ピリミジン-2-イル}アミノ)-2-メチルベンゼンスルホンアミド 一塩酸塩 |
| 分子式 | C21H23N7O2S・HCl |
| 分子量 | 473.98 |
| 物理化学的性状 | パゾパニブ塩酸塩は白色〜わずかに黄色の粉末である。 pH1.0の緩衝液に溶けにくく、pH7.0の緩衝液にほとんど溶けない。また、メタノールに溶けにくく、水又はエタノールに極めて溶けにくい。 |
| KEGG DRUG |
|
22. 包装
20錠[10錠(PTP)×2]
23. 主要文献
- NDBを用いた調査結果の概要(VEGF/VEGFR阻害作用を有する薬剤の動脈解離に関するリスク評価), (https://www.pmda.go.jp/files/000266521.pdf)
- Inada-Inoue,M.et al., Cancer Chemother.Pharmacol., 73, 673-683, (2014) »PubMed
- 社内資料:海外第I相試験(VEG10004)(2012年9月28日承認、CTD2.7.6 VEG10004試験)
- 社内資料:海外第I相試験(VEG10005)(2012年9月28日承認、CTD2.7.6 VEG10005試験)
- 社内資料:分布に関する試験(1)(2012年9月28日承認、CTD2.7.2.2.1.1)
- 社内資料:分布に関する試験(2)(2012年9月28日承認、CTD2.7.2.2.1.2)
- 社内資料:分布に関する試験(3)(2012年9月28日承認、CTD2.6.4.4.6)
- 社内資料:分布に関する試験(4)(2012年9月28日承認、CTD2.6.4.4.7)
- 社内資料:代謝に関する試験(1)(2012年9月28日承認、CTD2.7.2.2.1.4)
- 社内資料:海外第I,II及びIII相試験(母集団薬物動態)(2012年9月28日承認、CTD2.7.2.3.2)
- 社内資料:海外第I相試験(NCI-8063)(2012年9月28日承認、CTD2.7.6 NCI-8063試験)
- 社内資料:相互作用に関する試験(1)(2012年9月28日承認、CTD2.6.4.5.5.1.1)
- 社内資料:海外第I相試験(VEG10007)(2012年9月28日承認、CTD2.7.6 VEG10007試験)
- 社内資料:相互作用に関する試験(2)(2012年9月28日承認、CTD2.6.4.4.10)
- 社内資料:国際共同第III相試験(VEG110727)(2012年9月28日承認、CTD2.7.6 VEG110727試験)
- 社内資料:海外第II相試験(VEG20002)(2012年9月28日承認、CTD2.7.6 VEG20002試験)
- van Glabbeke,M.et al., Eur.J.Cancer., 38, 543-549, (2002) »PubMed
- 社内資料:国際共同第III相試験(VEG108844)(2014年3月17日承認、CTD2.7.6 VEG108844試験)
- 社内資料:海外第III相試験(VEG105192)(2014年3月17日承認、CTD2.7.6 VEG105192試験)
- Kumar,R.et al., Mol.Cancer Ther., 6, 2012-2021, (2007) »PubMed
- Hosaka,S.et al., J.Orthop.Res., 30, 1493-1498, (2012) »PubMed
- 社内資料:薬効薬理試験(1)(2012年9月28日承認、CTD2.6.2.2.2.3.1)
- 社内資料:薬効薬理試験(2)(2012年9月28日承認、CTD2.6.2.2.2.3.2)
24. 文献請求先及び問い合わせ先
文献請求先
ノバルティスファーマ株式会社
ノバルティスダイレクト
〒105-6333
東京都港区虎ノ門1-23-1
電話:0120-003-293 受付時間:月〜金 9:00〜17:30(祝祭日及び当社休日を除く)
製品情報問い合わせ先
ノバルティスファーマ株式会社
ノバルティスダイレクト
〒105-6333
東京都港区虎ノ門1-23-1
電話:0120-003-293 受付時間:月〜金 9:00〜17:30(祝祭日及び当社休日を除く)
26. 製造販売業者等
26.1 製造販売
ノバルティスファーマ株式会社
東京都港区虎ノ門1-23-1