医薬品情報
| 総称名 | ザガーロ |
|---|---|
| 一般名 | デュタステリド |
| 欧文一般名 | Dutasteride |
| 製剤名 | デュタステリドカプセル |
| 薬効分類名 | 5α還元酵素1型/2型阻害薬 男性型脱毛症治療薬 |
| 薬効分類番号 | 2499 |
| ATCコード | G04CB02 |
| KEGG DRUG |
D03820
デュタステリド
|
| JAPIC | 添付文書(PDF) |
添付文書情報2025年8月 改訂(第2版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ザガーロカプセル0.1mg | ZAGALLO Capsules | グラクソ・スミスクライン | 249900AM1023 | 劇薬, 処方箋医薬品注) | |
| ザガーロカプセル0.5mg | ZAGALLO Capsules | グラクソ・スミスクライン | 249900AM2020 | 劇薬, 処方箋医薬品注) |
2. 禁忌
次の患者には投与しないこと
4. 効能または効果
男性における男性型脱毛症
5. 効能または効果に関連する注意
5.1 男性における男性型脱毛症のみの適応である。他の脱毛症に対する適応はない。
5.2 20歳未満での安全性及び有効性は確立されていない。
6. 用法及び用量
男性成人には、通常、デュタステリドとして0.1mgを1日1回経口投与する。なお、必要に応じて0.5mgを1日1回経口投与する。
7. 用法及び用量に関連する注意
7.1 投与開始後12週間で改善が認められる場合もあるが、治療効果を評価するためには、通常6ヵ月間の治療が必要である。
7.2 本剤を6ヵ月以上投与しても男性型脱毛症の改善がみられない場合には投薬を中止すること。また、6ヵ月以上投与する場合であっても定期的に効果を確認し、継続投与の必要性について検討すること。
8. 重要な基本的注意
8.2 本剤は、血清前立腺特異抗原(PSA)に影響を与えるので、前立腺癌等の検査に際しては、以下の点に注意すること。また、PSAの検査を受ける際には本剤の服用について検査を行う医師に知らせるよう、患者を指導すること。
・PSA値は、前立腺癌のスクリーニングにおける重要な指標である。一般に、PSA値が基準値(通常、4.0ng/mL)以上の場合には、更なる評価が必要となり、前立腺生検の実施を考慮に入れる必要がある。なお、本剤投与中の患者で、本剤投与前のPSA値が基準値未満であっても、前立腺癌の診断を除外しないように注意すること。
・本剤投与6ヵ月以降のPSA値を新たなベースラインとし、その後は適宜PSA値を測定してベースラインからの変動を評価すること。
・デュタステリドは、前立腺肥大症患者に0.5mg/日投与した場合、前立腺癌の存在下であっても、投与6ヵ月後にPSA値を約50%減少させる。したがって、本剤を6ヵ月以上投与している患者のPSA値を評価する際には、測定値を2倍した値を目安として基準値と比較すること。また、PSA値は、本剤投与中止後6ヵ月以内に本剤投与開始前の値に戻る。なお、男性型脱毛症患者においても、臨床試験の結果から、本剤投与によりPSA値が減少すると推測される。
・本剤投与中におけるPSA値の持続的増加に対しては、前立腺癌の発現や本剤の服薬不遵守を考慮に含め、注意して評価すること。
・本剤投与中において、free/total PSA比は一定に維持されるので、前立腺癌のスクリーニングの目的で% free PSAを使用する場合には、測定値の調整は不要である。
9. 特定の背景を有する患者に関する注意
9.3 肝機能障害患者
9.3.1 重度の肝機能障害のある患者
投与しないこと。本剤は主に肝臓で代謝されるため、血中濃度が上昇するおそれがある。[2.4参照]
9.3.2 肝機能障害のある患者(重度の肝機能障害のある患者を除く)
本剤は主に肝臓で代謝される。肝機能障害のある患者に投与した場合の薬物動態は検討されていない。[16.4.1参照]
9.5 妊婦
9.6 授乳婦
9.7 小児等
10. 相互作用
相互作用序文
本剤は、主としてCYP3A4で代謝される。[16.4.1参照]
薬物代謝酵素用語
CYP3A4
10.2 併用注意
| CYP3A4阻害作用を有する薬剤 リトナビル等 [16.7.2参照] | これらの薬剤との併用により本剤の血中濃度が上昇する可能性がある。 | CYP3A4による本剤の代謝が阻害される。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
肝機能障害、黄疸(いずれも頻度不明)
AST、ALT、ビリルビンの上昇等を伴う肝機能障害や黄疸があらわれることがある。
11.2 その他の副作用
| 1%以上 | 1%未満 | 頻度不明 | |
| 過敏症 | 発疹 | 蕁麻疹、アレルギー反応、そう痒症、限局性浮腫、血管性浮腫 | |
| 精神神経系 | 頭痛、抑うつ気分 | 浮動性めまい、味覚異常 | |
| 生殖系及び乳房障害 | 性機能不全(リビドー減退、勃起不全、射精障害)注) | 乳房障害(女性化乳房、乳頭痛、乳房痛、乳房不快感) | 精巣痛、精巣腫脹 |
| 皮膚 | 脱毛症(主に体毛脱落)、多毛症 | ||
| 消化器 | 腹部不快感 | 腹痛、下痢 | |
| その他 | 倦怠感、血中CK増加 |
14. 適用上の注意
14.1 薬剤交付時の注意
14.1.1 PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
14.1.2 カプセルの内容物が口腔咽頭粘膜を刺激する場合があるので、カプセルは噛んだり開けたりせずに服用させること。
15. その他の注意
15.1 臨床使用に基づく情報
15.1.1 海外臨床試験において、18〜52歳の健康成人(デュタステリド群:27例、プラセボ群:23例)を対象に、52週間の投与期間及び24週間の投与後追跡期間を通して、デュタステリド0.5mg/日の精液特性に対する影響を評価した。投与52週目における総精子数、精液量及び精子運動率の投与前値からの平均減少率(プラセボ群の投与前値からの変化で調整)は、それぞれ23、26及び18%であり、精子濃度及び精子形態への影響は認められなかった。デュタステリド群における総精子数の投与前値からの平均減少率は、24週間の追跡期間後においても23%のままであった。しかしながら、いずれの評価時期においても、全ての精液パラメータの平均値は正常範囲内であり、事前に規定した臨床的に重要な変動(30%)には至らなかった。また、デュタステリド群の2例において、投与52週目に投与前値から90%を超える精子数の減少が認められたが、追跡24週目には軽快した。デュタステリドの精液特性に及ぼす影響が、個々の患者の受胎能に対しどのような臨床的意義をもつかは不明である。
15.1.2 デュタステリドを投与された前立腺肥大症患者で男性乳癌が報告されている。デュタステリドと男性乳癌の発現との関連性は不明である。なお、前立腺肥大症患者を対象とした2〜4年間の海外臨床試験(4325例)において3例の乳癌が報告された。このうち、デュタステリドが投与された症例では2例(曝露期間10週間、11ヵ月)、プラセボのみが投与された症例では1例報告されている。国内臨床試験での報告はない。
15.1.3 白人を主体とした50〜75歳の男性8231例(生検により前立腺癌が陰性かつPSA値2.5〜10.0ng/mL)を対象とした4年間の国際共同試験(日本人57例を含む)において、Modified Gleason Score注)8〜10の前立腺癌の発現率がプラセボ群(0.5%)に対しデュタステリド群(1.0%)において高かった(相対リスク2.06[95%信頼区間:1.13-3.75])との報告がある1)2)3)。
注)組織学的悪性度の指標
15.2 非臨床試験に基づく情報
15.2.1 アカゲザルの器官形成期にデュタステリドを2010ng/匹/日まで静脈内投与した結果、2010ng/匹/日群(デュタステリドを服用した男性の精液5mLを介して100%吸収されると仮定した場合に、体重50kgの女性が曝露される推定最大曝露量の186倍に相当する)の雌胎児1例に、本薬投与との関連性は不明であるが、卵巣・卵管の不均衡発達が認められた。
15.2.2 ラットのがん原性試験において、高用量(臨床用量における曝露量の約141倍)投与時に精巣間細胞腫の増加がみられた。しかしながら、精巣間細胞腫及び過形成の発現に起因するラットの内分泌機構のヒトへの外挿性が低いことから、ヒトに精巣間細胞腫を発現させる危険性は低いと考えられている。なお、マウスのがん原性試験においては、デュタステリドに関連すると考えられる腫瘍の発生は認められなかった。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
健康成人にデュタステリド0.5mgを単回経口投与した時、投与後1.5時間に最高血清中薬物濃度(Cmax平均値:3288.5pg/mL)に達し、AUC0-tは52316.9hr・pg/mL(平均値)であった(外国人データ:図1及び表1)。
図1 健康成人にデュタステリド0.5mgを単回経口投与した時の血清中薬物濃度(外国人データ)
(平均値+標準偏差、33例)
| Cmax(pg/mL) | AUC0-t(hr・pg/mL) | Tmax(hr) |
| 3288.5±1160.89 | 52316.9±20525.60 | 1.500(0.75-6.00) |
16.1.2 反復投与
男性の男性型脱毛症患者にデュタステリド0.05〜2.5mg注)を1日1回24週間反復経口投与した時、投与後24週の平均血清中薬物濃度は0.1及び0.5mg投与群でそれぞれ1.51±0.96及び30.69±13.90ng/mLであった。消失は非線形であり、血清中デュタステリド濃度が低い場合、高濃度域と比べて速やかに消失した(図2)。デュタステリド0.1及び0.5mgを24週間反復投与した時、血清中薬物濃度はそれぞれ最終投与後12及び20週時で定量下限(0.1ng/mL)未満であった(外国人データ)。
図2 男性の男性型脱毛症患者にデュタステリド0.05〜2.5mg注)を1日1回24週間経口投与後の血清中薬物濃度(外国人データ)
(平均値+標準偏差、34〜47例)
前立腺肥大症患者にデュタステリド0.5mgを1日1回6ヵ月間反復経口投与した時、投与後6ヵ月の血清中薬物濃度は44.82±17.91ng/mLであった。また、定常状態におけるt1/2は3.4±1.2週間であった。
16.2 吸収
16.2.1 食事の影響
健康成人にデュタステリド2.5mg注)を食後単回経口投与した時、薬物動態パラメータに若干の変化を認め、AUC0-∞は空腹時投与の2573から2197ng・hr/mLに減少した。なお、この変化は臨床上影響を与えるものではない。
16.2.2 生物学的利用率
健康成人にデュタステリド0.5mgを単回経口投与した時、生物学的利用率は59%であった(外国人データ)。
16.3 分布
16.3.1 蛋白結合率
In vitro試験において、デュタステリド(2000ng/mL)のヒト血清蛋白結合率は99.8%と高く、血清アルブミン、α1-酸性糖蛋白、コルチコステロイド結合グロブリン及び性ホルモン結合グロブリンに対する結合率は、それぞれ99.0%、96.6%、89.2%及び87.6%であった。また、これらの蛋白に対する結合率は20〜2000ng/mLの範囲で線形であった(限外ろ過法)。
16.3.2 精液移行
健康成人にデュタステリド0.5mgを反復経口投与した時、精液中/血清中薬物濃度比は平均11.5%であった(外国人データ)。
16.4 代謝
16.4.1 主な代謝酵素
16.4.2 代謝酵素阻害
In vitro試験において、デュタステリドはCYP1A2、2C9及び2D6活性を阻害しなかったが、CYP2C19及び3A4活性を阻害し、IC50は50μMであった。
16.4.3 代謝酵素誘導
In vitro試験において、デュタステリドはPXR活性化によるCYP3A4誘導能を示さなかった。
16.4.4 主代謝物
前立腺肥大症患者にデュタステリド0.5mgを1日1回反復経口投与した時、主代謝物として1,2-二水素化体、4'-水酸化体、6-水酸化体が確認された。
16.5 排泄
16.5.1 単回投与
健康成人にデュタステリド1〜20mg注)を単回経口投与した時、投与後48時間以内の尿中に未変化体は検出されなかった。
16.5.2 反復投与
健康成人にデュタステリド0.5mgを1日1回6ヵ月以上反復経口投与した時、糞中に約5%の未変化体が排泄され、関連物質(未変化体+代謝物)として約42%が回収された。尿中への未変化体の排泄は0.1%未満であり、関連物質の排泄も微量であった(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 高齢者
24〜87歳の健康成人にデュタステリド5mg注)を単回経口投与した時、50〜69歳及び70歳以上の年齢群のt1/2は49歳以下の年齢群に比べて延長し、AUC0-∞は約20%増加した。なお、この変化は臨床上影響を与えるものではない(外国人データ)。
16.7 薬物相互作用
16.7.1 トランスポーター阻害作用
In vitro試験において、デュタステリドはOAT3、OATP1B1及びOATP1B3輸送を阻害し、IC50の最小値はそれぞれ0.5、0.8及び20μMであったが、いずれも臨床血清中濃度(約0.07μM)より高かった。また、デュタステリドはMRP2及びOAT1輸送を阻害しなかった。
16.7.2 CYP3A4阻害作用を有する薬剤
(1)In vitro試験において、デュタステリドの酸化的代謝はCYP3A4阻害作用を有するケトコナゾールによって阻害された。[10.2参照]
(2)CYP3A4阻害薬とデュタステリドの薬物相互作用試験は実施されていないが、前立腺肥大症患者を対象とした臨床試験での母集団薬物動態解析の結果、ベラパミル塩酸塩又はジルチアゼム塩酸塩との併用により、デュタステリドのクリアランスが低下した(外国人データ)。[10.2参照]
16.7.3 その他の薬剤
デュタステリド0.5mgあるいは5mg注)と、コレスチラミン、ワルファリン、ジゴキシン、タムスロシン塩酸塩、テラゾシン塩酸塩との併用において薬物相互作用は認められなかった(外国人データ)。
注)本剤の承認用量は1日1回0.1又は0.5mgである。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国際共同第II/III相二重盲検比較試験
20歳から50歳の男性の男性型脱毛症患者(Norwood-Hamilton分類4)のIIIv、IV又はV:図1)917例(日本人200例を含む)を対象とし、本剤(0.02注)、0.1及び0.5mg)を24週間投与した際のプラセボ及びフィナステリド1mgに対する有効性及び安全性を検討した。その結果、頭頂部円内(直径2.54cm円中)の毛髪数のベースラインからの変化において、本剤0.1及び0.5mgのプラセボに対する優越性及びフィナステリド1mgに対する非劣性が検証された(表1及び図2)5)。
図1 臨床試験の対象となった脱毛タイプ(Norwood-Hamilton分類)
| プラセボ(181例) | デュタステリド | フィナステリド1mg(179例) | |||
| 0.02mg(185例) | 0.1mg(188例) | 0.5mg(184例) | |||
| 24週時 | |||||
| 例数 | 148 | 155 | 158 | 150 | 141 |
| 変化量(SE) | −4.9(7.89) | 17.1(7.74) | 63.0(7.67) | 89.6(7.87) | 56.5(8.12) |
| プラセボとの差(p値)注1) | − | 22.0(p=0.046) | 67.9(p<0.001) | 94.4(p<0.001) | 61.4(p<0.001) |
| フィナステリドとの差[99.165%信頼区間]注2) (p値)注1) | − | −39.4[−66.1,−12.7] (p<0.001) | 6.5[−20.1,33.1] (p=0.56) | 33.0[6.1,60.0] (p=0.003) | − |
図2 二重盲検比較試験:本剤(0.02注)、0.1及び0.5mg)の頭頂部円内(直径2.54cm円中)の毛髪数のベースラインからの変化量の推移
*プラセボとの優越性
♯フィナステリド1mgとの非劣性
副作用発現頻度(本剤0.02mg群を含む)は、17.1%(95/557例)であった。主な副作用は、勃起不全4.3%(24/557例)、リビドー減退3.9%(22/557例)、精液量減少1.3%(7/557例)であった。
17.1.2 国内第III相試験(長期投与試験)
20歳から50歳の男性の男性型脱毛症患者(Norwood-Hamilton分類4)のIIIv、IV又はV:図1)120例を対象とし、本剤0.5mgを52週間投与した際の安全性及び有効性を検討した。その結果、52週時の頭頂部円内(直径2.54cm円中)の毛髪数のベースラインからの変化量は、68.1本であり改善が示された。
副作用発現頻度は、16.7%(20/120例)であった。主な副作用は、勃起不全10.8%(13/120例)、リビドー減退8.3%(10/120例)、射精障害4.2%(5/120例)であった。
副作用発現頻度は、16.7%(20/120例)であった。主な副作用は、勃起不全10.8%(13/120例)、リビドー減退8.3%(10/120例)、射精障害4.2%(5/120例)であった。
注)本剤の承認用量は1日1回0.1又は0.5mgである。
18. 薬効薬理
18.1 作用機序
デュタステリドは、テストステロンをジヒドロテストステロンへ変換する1型及び2型5α還元酵素を阻害する。ジヒドロテストステロンは男性型脱毛症に関与する主なアンドロゲンである。
18.2 5α還元酵素阻害作用
In vitroにおいて、ヒト1型及び2型5α還元酵素を阻害した6)。
18.3 血清中のジヒドロテストステロン濃度低下作用
男性の男性型脱毛症患者に本剤0.1及び0.5mgを1日1回24週間反復経口投与した時の結果を下表に示す。
| 評価時点 | プラセボ | デュタステリド | |
| 0.1mg | 0.5mg | ||
| 12週時 | −2.6% | −85.8% | −91.2%注) |
| 24週時 | −6.2% | −83.6% | −90.9%注) |
18.4 頭皮中のジヒドロテストステロン濃度低下作用
男性の男性型脱毛症患者に本剤0.1及び0.5mgを1日1回反復経口投与した時、投与6ヵ月のジヒドロテストステロン濃度はベースラインからそれぞれ血清中で65及び90%減少し、頭皮中で40及び52%減少した(調整済み平均値)。また、本剤投与による頭皮中ジヒドロテストステロン濃度の低下と発毛作用(毛髪数のベースラインからの増加量)との間には関連性がみられた(外国人データ)。
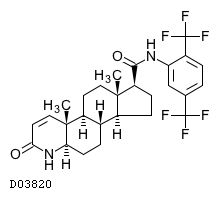
19. 有効成分に関する理化学的知見
19.1. デュタステリド
| 一般的名称 | デュタステリド |
|---|---|
| 一般的名称(欧名) | Dutasteride |
| 化学名 | N-[2,5-Bis(trifluoromethyl)phenyl]-3-oxo-4-aza-5α-androst-1-ene-17β-carboxamide |
| 分子式 | C27H30F6N2O2 |
| 分子量 | 528.53 |
| 融点 | 242〜250℃ |
| 物理化学的性状 | 白色〜微黄色の粉末である。 |
| 分配係数 | (logP):4.9(1-オクタノール/水系) |
| KEGG DRUG |
|
20. 取扱い上の注意
光及び湿気を避けるため、PTP包装のまま保存すること。
22. 包装
<ザガーロカプセル0.1mg>
30カプセル[10カプセル(PTP)×3]
<ザガーロカプセル0.5mg>
30カプセル[10カプセル(PTP)×3]
23. 主要文献
- Andriole GL,et al., N Engl J Med., 362, 1192-1202, (2010) »PubMed
- Theoret MR,et al., N Engl J Med., 365, 97-99, (2011) »PubMed
- Akaza H,et al., Jpn J Clin Oncol., 41, 417-423, (2011) »PubMed
- Norwood OT,et al., South Med J., 68, 1359-1365, (1975) »PubMed
- Gubelin HW,et al., J Am Acad Dermatol., 70, 489-498, (2014)
- Tian G,et al., Biochemistry., 34, 13453-13459, (1995) »PubMed
24. 文献請求先及び問い合わせ先
文献請求先
グラクソ・スミスクライン株式会社
メディカル・インフォメーション
東京都港区赤坂1-8-1
電話:0120-561-007(9:00〜17:45/土日祝日及び当社休業日を除く)
製品情報問い合わせ先
グラクソ・スミスクライン株式会社
メディカル・インフォメーション
東京都港区赤坂1-8-1
電話:0120-561-007(9:00〜17:45/土日祝日及び当社休業日を除く)
25. 保険給付上の注意
本剤は保険給付の対象とならない(薬価基準未収載)。
26. 製造販売業者等
26.1 製造販売元
グラクソ・スミスクライン株式会社
東京都港区赤坂1-8-1