医薬品情報
| 総称名 | カプレルサ |
|---|---|
| 一般名 | バンデタニブ |
| 欧文一般名 | Vandetanib |
| 製剤名 | バンデタニブ製剤 |
| 薬効分類名 | 抗悪性腫瘍剤 チロシンキナーゼ阻害剤 |
| 薬効分類番号 | 4291 |
| ATCコード | L01EX04 |
| KEGG DRUG |
D06407
バンデタニブ
|
| JAPIC | 添付文書(PDF) |
添付文書情報2025年11月 改訂(第3版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| カプレルサ錠100mg | Caprelsa Tablets | サノフィ | 4291041F1029 | 7902.2円/錠 | 劇薬, 処方箋医薬品 |
1. 警告
2. 禁忌
次の患者には投与しないこと
4. 効能または効果
根治切除不能な甲状腺髄様癌
6. 用法及び用量
通常、成人にはバンデタニブとして1回300mgを1日1回、経口投与する。なお、患者の状態により適宜減量する。
7. 用法及び用量に関連する注意
7.1 副作用により本剤を減量、休薬又は中止する場合には、副作用の症状、重症度に応じて以下の基準を考慮すること。[1.3、8.2、11.1.2参照]
| 休薬・減量基準 | 投与量調節 | |
| QT間隔延長 | 500msecを超えるQTcB | QTcBが480msec以下に軽快するまで本剤を休薬し、再開する場合には休薬前の投与量から減量すること。本剤を休薬し、6週間以内に480msec以下に軽快しない場合には、本剤の投与を中止すること。 |
| その他の副作用 | グレード3以上 | 回復又はグレード1に軽快するまで本剤を休薬し、再開する場合には休薬前の投与量から減量すること。 |
7.2 本剤を減量する場合には、1日1回200mgに減量し、その後必要であれば100mgに減量すること。
7.3 本剤と他の抗悪性腫瘍剤との併用について、有効性及び安全性は確立していない。
8. 重要な基本的注意
8.1 間質性肺疾患があらわれることがあるので、初期症状(息切れ、呼吸困難、咳嗽、発熱等)の確認、定期的な胸部画像検査の実施等、患者の状態を十分に観察すること。また、必要に応じて動脈血酸素分圧(PaO2)、動脈血酸素飽和度(SpO2)、肺胞気動脈血酸素分圧較差(A-aDO2)、肺拡散能力(DLco)等の検査を行うこと。[1.2、9.1.1、11.1.1参照]
8.2 QT間隔延長があらわれることがあるので、投与開始前及び投与中は定期的に心電図検査及び電解質検査(カリウム、マグネシウム、カルシウム等)を行い、患者の状態を十分に観察すること。また、必要に応じて電解質を補正すること。[1.3、2.2、7.1、9.1.2、10.2、11.1.2参照]
8.3 重篤な心障害があらわれることがあるので、投与開始前及び投与中はこれらの症状の発現状況・重篤度等に応じて適宜心機能検査(心エコー等)を行い、患者の状態を十分に観察すること。[9.1.3、11.1.3参照]
8.5 肝障害があらわれることがあるので、投与中は定期的に肝機能検査を行い、患者の状態を十分に観察すること。[11.1.11参照]
8.6 手足症候群、中毒性表皮壊死融解症(Toxic Epidermal Necrolysis:TEN)、皮膚粘膜眼症候群(Stevens-Johnson症候群)、多形紅斑等の皮膚障害があらわれることがあるので、患者の状態を十分に観察すること。また、必要に応じて皮膚科を受診するよう、患者に指導すること。[11.1.5参照]
8.7 創傷治癒を遅らせる可能性があるので、外科的処置が予定されている場合には、外科的処置の前に本剤の投与を中断すること。外科的処置後の投与再開は、患者の状態に応じて判断すること。
8.8 霧視等の重篤な眼障害があらわれることがあるので、投与中は定期的に眼の異常の有無を確認すること。異常が認められた場合には、速やかに医療機関を受診するよう患者を指導すること。
8.9 疲労、霧視等があらわれることがあるので、自動車の運転等、危険を伴う機械の操作に従事する際には注意するよう患者に十分に説明すること。
8.10 定期的に血清カルシウム濃度を測定すること。[11.1.10参照]
8.11 定期的に甲状腺刺激ホルモン濃度を測定すること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 間質性肺疾患のある患者又はその既往歴のある患者
9.1.2 QT間隔延長のおそれ又はその既往歴のある患者
9.1.3 心不全症状のある患者又はその既往歴のある患者
9.1.4 高血圧症の患者
9.2 腎機能障害患者
本剤の減量を考慮するとともに、患者の状態をより慎重に観察し、有害事象の発現に十分注意すること。本剤の血中濃度が上昇することが報告されている。[16.6.1参照]
9.4 生殖能を有する者
妊娠する可能性のある女性には、本剤投与中及び最終投与後4カ月間において避妊する必要性及び適切な避妊法について説明すること。[9.5参照]
9.5 妊婦
9.6 授乳婦
授乳しないことが望ましい。動物実験(ラット)で乳汁中へ移行することが報告されている。
9.7 小児等
小児等を対象とした臨床試験は実施していない。
9.8 高齢者
患者の状態を観察しながら慎重に投与すること。一般に、生理機能が低下していることが多い。
10. 相互作用
相互作用序文
本剤はCYP3A4の基質となる。また、本剤は有機カチオントランスポーター2(OCT2)及びP-糖蛋白を阻害することが示されている。
薬物代謝酵素用語
CYP3A4
薬物代謝酵素用語
有機カチオントランスポーター2(OCT2)
薬物代謝酵素用語
P-糖蛋白
10.2 併用注意
| 抗不整脈剤 キニジン プロカインアミド ジソピラミド等 QT間隔延長を起こすおそれがある他の薬剤 オンダンセトロン クラリスロマイシン ハロペリドール等 [1.3、2.2、8.2、9.1.2、11.1.2参照] | QT間隔延長を起こす又は悪化させるおそれがあるので、QT間隔延長を起こすことが知られている薬剤と併用する場合には、治療上の有益性が危険性を上回ると判断される場合にのみ使用すること。 | 本剤及びこれらの薬剤はいずれもQT間隔を延長させるおそれがあるため、併用により作用が増強するおそれがある。 |
| CYP3A誘導剤 フェニトイン カルバマゼピン リファンピシン バルビツール酸系薬物 セイヨウオトギリソウ(St.John's Wort、セント・ジョーンズ・ワート)含有食品等 [16.7.1参照] | CYP3A誘導剤との併用により、本剤の血漿中濃度が低下するおそれがある。 | 本剤の代謝には主にCYP3A4が関与しているため、左記薬剤のようなCYP3A誘導剤との併用で、本剤の代謝が亢進し血漿中濃度が低下する可能性がある。 |
| OCT2の基質となる薬剤 メトホルミン等 [16.7.2参照] | OCT2基質との併用により、OCT2基質の血漿中濃度が上昇するおそれがある。 | 本剤はOCT2の阻害剤であるため、OCT2基質との併用によりOCT2基質の血漿中濃度が増加する可能性がある。 |
| P-糖蛋白の基質となる薬剤 ジゴキシン アリスキレン フェキソフェナジン サキサグリプチン シタグリプチン等 [16.7.3参照] | P-糖蛋白基質との併用により、P-糖蛋白基質の血漿中濃度が上昇するおそれがある。 | 本剤はP-糖蛋白の阻害剤であることから、本剤とP-糖蛋白基質との併用によりP-糖蛋白基質の血漿中濃度が増加する可能性がある。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 間質性肺疾患(0.4%)
11.1.3 心障害(6.1%)
11.1.4 重度の下痢(9.4%)
脱水、電解質異常等の異常が認められた場合には、本剤の休薬、減量又は中止等の適切な処置を行うこと。
11.1.5 中毒性表皮壊死融解症(Toxic Epidermal Necrolysis:TEN)(頻度不明)、皮膚粘膜眼症候群(Stevens-Johnson症候群)(頻度不明)、多形紅斑(頻度不明)[8.6参照]
11.1.6 重度の皮膚障害(20.4%)
光線過敏反応、発疹、皮膚潰瘍等の重度の皮膚障害があらわれることがある。
11.1.7 高血圧
11.1.8 可逆性後白質脳症症候群(頻度不明)
痙攣、頭痛、視覚障害、錯乱、皮質盲等が認められた場合には投与を中止し、血圧のコントロール等の適切な処置を行うこと。
11.1.9 腎障害
腎不全(0.4%)、蛋白尿(9.8%)等があらわれることがある。
11.1.10 低カルシウム血症(6.1%)
異常が認められた場合には、カルシウム剤やビタミンD製剤の投与等の適切な処置を行うこと。[8.10参照]
11.1.11 肝障害
ALT増加(3.3%)、AST増加(3.7%)、血中ビリルビン増加(頻度不明)等があらわれることがある。[8.5参照]
11.1.12 出血
鼻出血(4.9%)、血尿(0.4%)、くも膜下出血(頻度不明)等があらわれることがある。
11.1.13 消化管穿孔
小腸穿孔(0.4%)等があらわれることがある。
11.1.14 動脈解離(頻度不明)
大動脈解離を含む動脈解離があらわれることがある1)。
11.2 その他の副作用
| 10%以上 | 1〜10%未満 | 1%未満 | 頻度不明 | |
| 皮膚 | 皮膚症状(発疹、ざ瘡、皮膚乾燥、皮膚炎、そう痒症等) | 手掌・足底発赤知覚不全症候群、脱毛症、爪の障害 | 長睫毛症、擦過傷、メラノサイト性母斑、毛髪成長異常、毛質異常、多汗症、寝汗 | |
| 消化器 | 下痢、悪心、食欲減退 | 消化不良、嘔吐、腹痛、便秘、嚥下障害、口内炎、口内乾燥 | 膵炎、腹部膨満、唾液欠乏、放屁、胃腸音異常 | |
| 呼吸器 | 咳嗽、呼吸困難、発声障害 | 鼻乾燥 | ||
| 筋・骨格系及び結合組織 | 無力症、関節炎、筋骨格系胸痛、筋痙縮 | 筋力低下 | 骨壊死 | |
| 血液 | ヘモグロビン増加、リンパ球減少症 | 貧血 | ||
| 内分泌 | 甲状腺機能低下症 | |||
| 精神神経系 | 頭痛、睡眠障害(不眠症、嗜眠等)、うつ病、味覚異常、聴力低下、ニューロパチー、めまい、錯感覚、振戦、神経過敏、注意力障害、不安、性欲減退 | 口の感覚鈍麻、知覚過敏、感覚鈍麻 | ||
| 眼 | 角膜混濁 | 結膜炎、眼乾燥、視力障害、霧視 | 眼の障害、眼瞼浮腫、緑内障、羞明、光視症、マイボーム腺機能不全 | |
| その他 | 疲労 | 体重減少、脱水、体重増加、疼痛、ほてり、潮紅、全身健康状態低下、低カリウム血症、低マグネシウム血症、尿意切迫、発熱、浮腫 | 虚血性脳血管障害、狭心症、治癒不良、粘膜の炎症、低ナトリウム血症、意識消失、頻尿、末梢冷感 |
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
15. その他の注意
15.2 非臨床試験に基づく情報
ラット反復投与毒性試験において、ヒトにおける曝露量よりも低い曝露量で、肺、肝臓、腎臓、脾臓等にリン脂質症に関連する所見(ミエリンの渦状形成による細胞質の空胞化)が認められた。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
日本人固形癌患者18例において本剤100、200、300及び400mg注4)を単回経口投与したとき、最高血漿中濃度は投与4〜6時間後に認められ、その後、血漿中濃度は2相性の消失を示した。Cmax及びAUC0-∞は100〜400mgの用量範囲で、ほぼ用量に比例して増加した2)。
| 用量注4)(mg/日) | 症例数 | Cmax(ng/mL) | tmax注1)(h) | AUC0-∞(μg・h/mL) | t1/2(h) |
| 100 | 3 | 103±42.0 | 6 | 10.1±3.53 | 115±46.0 |
| 200 | 6 | 186±91.6 | 4 | 16.8±6.94 | 101±14.1 |
| 300 | 6 | 392±198 | 5 | 29.4±11.8 | 90.2±13.7 |
| 400 | 3 | 447±240 | 6 | 32.1±4.66 | 114±44.7 |
バンデタニブ100、200、300及び400mg注4)を単回経口投与後の血漿中濃度推移(平均値±標準偏差)
16.1.2 反復投与
日本人固形癌患者において本剤100、200、300及び400mg注4)を1日1回28日間反復経口投与したとき、血漿中バンデタニブ濃度は投与開始後28日以降に定常状態に到達すると考えられた2)。
| 用量注4)(mg/日) | 症例数 | Cmax(ng/mL) | tmax注2)(h) | AUC0-24(μg・h/mL) | 累積係数注3) |
| 100 | 3 | 1200±583 | 4 | 20.5±5.00 | 14.2±1.8 |
| 200 | 4 | 922±259 | 6 | 18.3±5.71 | 6.2±1.9 |
| 300 | 3 | 1580±302 | 6 | 29.9±4.60 | 5.3±1.2 |
| 400 | 1 | 2050 | 4 | 44.6 | 6.5 |
16.2 吸収
16.2.1 食事の影響
健康被験者16例を対象に、本剤300mgを食後に投与したとき、本剤のAUCには食事による影響は認められなかった。本剤のCmaxには、空腹時投与に比べ、食後投与で僅かな(11%)減少が認められた3)(外国人データ)。
16.3 分布
本薬はヒト血清アルブミン及びヒトα1-酸性糖蛋白に結合し、蛋白結合率は約90%である4)(in vitro)。
16.4 代謝
16.5 排泄
健康男性被験者4例に14C標識バンデタニブ800mgを単回経口投与したとき、投与後21日までの総放射能排泄率は約69%であった。糞及び尿中にはそれぞれ投与した放射能の約44%及び25%が排泄された3)(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害を有する被験者における薬物動態
クレアチニンクリアランス(CrCL)に基づく軽度(CrCL:50mL/min以上80mL/min以下)、中等度(CrCL:30mL/min以上50mL/min未満)及び重度(CrCL:30mL/min未満)の腎機能障害者、並びに健康被験者(CrCL:80mL/min超)の計32例を対象に、本剤800mg注4)を単回経口投与した。軽度、中等度及び重度腎機能障害者では、腎機能が正常な健康被験者に比べ、バンデタニブのAUCはそれぞれ46%(軽度)、62%(中等度)及び79%(重度)と高値を示した。一方、バンデタニブのCmaxは軽度、中等度及び重度腎機能障害者では、腎機能が正常な健康被験者に比べ、それぞれ7%(軽度)、9%(中等度)及び11%(重度)高値を示したが、明らかな差異は認められなかった7)(外国人データ)。[9.2参照]
16.6.2 肝機能障害を有する被験者における薬物動態
軽度(Child-Pugh分類A)、中等度(Child-Pugh分類B)及び重度(Child-Pugh分類C)の肝機能障害者並びに健康被験者の計30例に、本剤800mg注4)を単回経口投与した。バンデタニブのAUCには、健康被験者といずれの肝機能障害者との間で差は認められなかった。一方、バンデタニブのCmaxには、健康被験者と軽度あるいは中等度肝機能障害者の間に差は認められなかったものの、健康被験者に比べ、重度肝機能障害者ではCmaxは29%低かった8)(外国人データ)。
16.7 薬物相互作用
16.7.1 リファンピシン(CYP3A誘導剤)との併用による影響
16.7.2 メトホルミン(OCT2基質)との併用による影響
16.7.3 ジゴキシン(P-糖蛋白基質)との併用による影響
16.7.4 イトラコナゾール(CYP3A阻害剤)との併用による影響
健康被験者15例を対象に、バンデタニブ300mg及びイトラコナゾール200mg/日を併用投与したとき、バンデタニブ単独投与時に比べ、バンデタニブのAUCは9%増加した。バンデタニブのCmaxには、イトラコナゾール併用による影響は認められなかった9)(外国人データ)。
16.7.5 ミダゾラム(CYP3A基質)との併用による影響
健康被験者17例を対象に、バンデタニブ800mg注4)及びミダゾラム7.5mgを併用投与したとき、バンデタニブはミダゾラムの曝露量に影響を及ぼさなかった10)(外国人データ)。
16.7.6 CYP1A2及び2C9に対するバンデタニブの誘導作用
CYP1A2及び2C9に対するバンデタニブの誘導作用を評価した結果、バンデタニブはCYP1A2及び2C9を誘導することが示された(in vitro)。CYP1A2及びCYP2C9に対する誘導作用は、陽性対照で認められた誘導作用のそれぞれ28%及び38%(いずれも最大値)である11)。
16.7.7 乳癌耐性タンパク(BCRP)に対するバンデタニブの阻害作用
バンデタニブはBCRPを若干阻害する(IC50値:11.9μg/mL)ことが示された12)(in vitro)。
注4)本剤の承認用量は300mg/日である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国内第1/2相試験
根治切除不能な甲状腺髄様癌の日本人患者14例を対象として、本剤300mg/日注1)の安全性及び忍容性を検討する非盲検による第1/2相試験を実施した。有効性解析対象集団13例における客観的奏効率は38.5%(5/13例)であった。副作用は100%(14例全例)に認められ、主な副作用は、下痢71.4%(10例)、高血圧64.3%(9例)及び発疹42.9%(6例)であった13)。
17.1.2 海外第3相試験
根治切除不能な甲状腺髄様癌患者(本剤群231例、プラセボ群100例)を対象として、本剤300mg/日の有効性及び安全性をプラセボと比較する二重盲検無作為化比較第3相試験を実施した。主要評価項目である画像中央判定に基づく無増悪生存期間の最終解析結果(中央値[95%信頼区間])は、本剤群でNE注2)[24.9〜NE]カ月、プラセボ群で19.3[15.1〜NE]カ月であり、本剤はプラセボに対し統計学的に有意な延長を示した(ハザード比0.46、95%信頼区間0.31〜0.69、p=0.0001[ログランク検定]、2009年7月31日データカットオフ)。副作用は本剤群96.1%(222/231例)に認められ、主な副作用は、下痢46.8%(108例)、発疹42.4%(98例)、高血圧24.7%(57例)、悪心23.4%(54例)、ざ瘡18.6%(43例)及び疲労18.6%(43例)であった14)15)。
無増悪生存期間のKaplan-Meier曲線(最大解析対象集団)
17.3 その他
17.3.1 QT間隔に及ぼす影響
健康被験者24例を対象に、バンデタニブ700mg注3)を単独投与したときQT間隔の延長(11.4ms)が認められた。また、バンデタニブと5HT3拮抗薬であるオンダンセトロン32mgを併用投与したとき、バンデタニブ単独投与に比べてさらにQT間隔が延長(10.8ms)することが示された16)(外国人データ)。
注1)中等度腎機能障害患者では200mg/日
注2)Not Estimable(推定不可)
注3)本剤の承認用量は300mg/日である。
18. 薬効薬理
18.1 作用機序
18.1.1 ヒト甲状腺髄様癌由来細胞株のVEGFR-2、EGFR、RET等のチロシンキナーゼのリン酸化を阻害することにより、細胞増殖を抑制した17)。
18.1.3 ヒト肺癌由来Calu-6細胞株を皮下移植したヌードマウスにおいて、バンデタニブによる腫瘍組織内の血管内皮細胞減少及び腫瘍細胞壊死の増加が認められた。ヒト肺癌由来A549細胞株を皮内移植したヌードマウスにおいて、バンデタニブによる腫瘍血管新生阻害が認められた19)。
18.2 抗腫瘍効果
19. 有効成分に関する理化学的知見
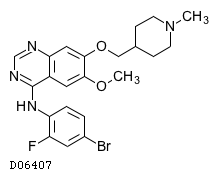
19.1. バンデタニブ
| 一般的名称 | バンデタニブ |
|---|---|
| 一般的名称(欧名) | Vandetanib |
| 化学名 | N-(4-bromo-2-fluorophenyl)-6-methoxy-7-[(1-methylpiperidin-4-yl)methoxy]quinazolin-4-amine |
| 分子式 | C22H24BrFN4O2 |
| 分子量 | 475.35 |
| 物理化学的性状 | 本品は白色の粉末である。 |
| KEGG DRUG |
|
21. 承認条件
21.1 医薬品リスク管理計画を策定の上、適切に実施すること。
21.2 国内での治験症例が極めて限られていることから、製造販売後、一定数の症例に係るデータが集積されるまでの間は、全症例を対象に使用成績調査を実施することにより、本剤使用患者の背景情報を把握するとともに、本剤の安全性及び有効性に関するデータを早期に収集し、本剤の適正使用に必要な措置を講じること。
22. 包装
50錠[10錠(PTP)×5]
23. 主要文献
- NDBを用いた調査結果の概要(VEGF/VEGFR阻害作用を有する薬剤の動脈解離に関するリスク評価), (https://www.pmda.go.jp/files/000266521.pdf)
- Tamura T,et al., J Thorac Oncol., 1 (9), 1002-9, (2006) »PubMed
- Martin P,et al., Clin Ther., 34 (1), 221-37, (2012) »PubMed
- 社内資料:血漿蛋白結合率(2015年9月28日承認、CTD2.7.2.2), (2000)
- 社内資料:チトクロームP450による代謝(2015年9月28日承認、CTD2.7.2.2), (2004)
- 社内資料:フラビン含有モノオキシゲナーゼによる代謝(2015年9月28日承認、CTD2.7.2.2), (2004)
- 社内資料:腎機能障害者における薬物動態試験(2015年9月28日承認、CTD2.7.2.3), (2009)
- 社内資料:肝機能障害者における薬物動態試験(2015年9月28日承認、CTD2.7.2.3), (2009)
- Martin P,et al., Drugs R D., 11 (1), 37-51, (2011) »PubMed
- Johansson S,et al., Clin Pharmacokinet., 53, 837-47, (2014) »PubMed
- 社内資料:チトクロームP450に及ぼす本薬の酵素誘導作用(2015年9月28日承認、CTD2.7.2.3), (2007)
- 社内資料:各種トランスポーターに及ぼす本薬の阻害作用(2015年9月28日承認、CTD2.7.2.2), (2008)
- 社内資料:甲状腺髄様癌患者を対象とした国内第1/2相臨床試験(2015年9月28日承認、CTD2.7.6.2)
- Wells SA,et al., J Clin Oncol., 30 (2), 134-41, (2012) »PubMed
- 社内資料:甲状腺髄様癌患者を対象とした海外第3相臨床試験(2015年9月28日承認、CTD2.7.6.2)
- 社内資料:本薬及びオンダンセトロンによる心筋の再分極に対する薬力学的作用(2015年9月28日承認、CTD2.7.2.3), (2004)
- Vitagliano D,et al., Endocrine-Related Cancer., 18, 1-11, (2011) »PubMed
- Brave SR,et al., Int J Oncol., 39, 271-8, (2011) »PubMed
- Wedge SR,et al., Cancer Res., 62, 4645-55, (2002) »PubMed
24. 文献請求先及び問い合わせ先
文献請求先
サノフィ株式会社
くすり相談室
〒163-1488
東京都新宿区西新宿三丁目20番2号
電話:フリーダイヤル 0120-109-905
製品情報問い合わせ先
サノフィ株式会社
くすり相談室
〒163-1488
東京都新宿区西新宿三丁目20番2号
電話:フリーダイヤル 0120-109-905
26. 製造販売業者等
26.1 製造販売元
サノフィ株式会社
〒163-1488
東京都新宿区西新宿三丁目20番2号