医薬品情報
| 総称名 | サブリル |
|---|---|
| 一般名 | ビガバトリン |
| 欧文一般名 | Vigabatrin |
| 製剤名 | ビガバトリン製剤 |
| 薬効分類名 | 抗てんかん剤 |
| 薬効分類番号 | 1139 |
| ATCコード | N03AG04 |
| KEGG DRUG |
D00535
ビガバトリン
|
| JAPIC | 添付文書(PDF) |
添付文書情報2023年7月 改訂(第1版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| サブリル散分包500mg | Sabril powder | サノフィ | 1139013B1026 | 1514.5円/包 | 劇薬, 処方箋医薬品 |
1. 警告
1.1 本剤の投与を受けた約1/3の患者で不可逆的な視野狭窄が起こることが報告されている注1)。本剤の投与は、点頭てんかんの診断、治療に精通し、かつ本剤の安全性及び有効性についての十分な知識を有し、サブリル処方登録システム(Sabril Registration System for Prescription:SRSP)注2)に登録された医師・薬剤師がおり、網膜電図検査などの眼科検査に精通した眼科専門医と連携が可能な登録医療機関において、登録患者に対してのみ行うこと。[1.2、2.2、8.1、9.1.1、11.1.1参照]
1.2 本剤による視野狭窄の発現頻度は曝露期間の延長、累積投与量の増加に伴い高くなるため、本剤投与開始時及び本剤投与中はSRSPに準拠して定期的に視野検査を含めた眼科検査を実施すること。視野狭窄、あるいは網膜電図検査などで異常が認められた場合は、本剤による治療の継続の必要性を慎重に判断し、治療上の有益性が危険性を上回る場合にのみ本剤による治療を継続すること。治療を継続する場合には、より頻回に眼科検査を行い、本剤による治療の継続が適切であるかどうか定期的に判断すること。[1.1、2.2、8.1、9.1.1、11.1.1参照]
1.3 本剤の投与にあたっては、患者又は代諾者に本剤の有効性及び危険性について文書によって説明し、文書で同意を取得すること。
注1)外国人の成人及び小児の難治性てんかん患者を対象とした試験において、成人では36.5%(110/301例)、小児では20.0%(17/85例)に1回以上の両側性の求心性視野狭窄がみられた。
注2)定期的な眼科検査を実施し、視野障害、視力障害の早期発見を目的として規定された手順
2. 禁忌
4. 効能または効果
6. 用法及び用量
通常、生後4週以上の患者には、ビガバトリンとして1日50mg/kgから投与を開始する。患者の症状に応じて、3日以上の間隔をあけて1日投与量として50mg/kgを超えない範囲で漸増するが、1日最大投与量は150mg/kg又は3gのいずれか低い方を超えないこととし、いずれも1日2回に分け、用時溶解して経口投与する。
7. 用法及び用量に関連する注意
本剤の投与開始後2〜4週間に治療効果が認められない場合、あるいは最高投与量である150mg/kg/日を投与しても症状の改善が認められない場合には、本剤の投与中止を考慮すること。
8. 重要な基本的注意
8.1 本剤の投与により不可逆的な視野障害及び視力障害の発現が報告されている。本剤による視野障害は軽度から重度の両側性求心性視野狭窄であり、通常鼻側からあらわれ、ほとんどの場合は耳側視野より鼻側視野が広範に欠損する。本剤による視野障害は3ヵ月程度で急激に発現又は悪化することがあるため、本剤による視野障害をモニタリングするため、少なくとも3ヵ月に一度は視力検査、対座法による視野評価等を実施して患者の視機能について確認すること。また、網膜電図などによる視野検査を少なくとも投与開始時、投与3ヵ月、9ヵ月及び12ヵ月並びにそれ以降少なくとも6ヵ月ごとに実施すること。[1.1、1.2、2.2、9.1.1、11.1.1参照]
8.2 本剤の投与により視床、基底核、脳幹、小脳等において頭部MRI異常(T2強調画像高信号、拡散強調画像異常信号)の発現が報告されており、髄鞘内浮腫が認められているとする報告もある1)ことから、本剤投与開始時及び本剤投与期間中は定期的に頭部MRI検査を実施すること。異常が認められた場合には、関連する神経症状の有無などの患者の状態を慎重に観察し、本剤のベネフィット・リスクを評価した上で、本剤による治療継続の可否を判断すること。[11.1.6参照]
8.3 本剤の投与により顕著な鎮静、昏迷、錯乱、意識消失等の脳症症状があらわれるとの報告があるため、本剤投与期間中はこれらの症状の発現に注意すること。また、脳症症状が認められた症例の中には、急速な増量を行った患者、腎機能障害患者が含まれていたことから、これらの患者では特に注意すること。[11.1.5参照]
8.4 本剤の投与によりジストニア、ジスキネジア、筋緊張亢進、協調運動障害等の運動障害があらわれることがあり、これらの症状は頭部MRI異常を伴う場合があるため、症状が認められた場合には必要に応じて頭部MRI検査の実施を考慮すること。[11.1.6参照]
8.5 連用中における投与量の急激な減量あるいは投与中止により、発作の増悪又は重積状態があらわれることがあるので、投与を中止する場合には徐々に減量するなど慎重に行うこと。
8.6 本剤の投与により眠気、注意力・集中力・反射運動能力等の低下が起こることがあるので、代諾者に対し注意を与えること。また、患者に対し本剤投与中には危険を伴う機械操作や遊戯などを行わないよう十分に注意を与えること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 黄斑症、網膜症、緑内障又は視神経萎縮の既往又は合併症を有する患者
9.1.2 精神病性障害、うつ病、行動障害の既往歴のある患者
激越、うつ病、異常思考、妄想反応等の精神症状の発現が報告されている。
9.2 腎機能障害患者
腎機能障害患者では低い用量で反応する可能性があるため、低用量からの投与開始、又は投与間隔の調節を考慮すること。腎機能障害のある乳幼児における用量調節方法に関する情報は得られていない。脳症のリスクが増大するおそれがある。[16.6.1参照]
9.5 妊婦
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。ヒトにおいて乳汁中に移行することが報告されている。
9.7 小児等
低出生体重児及び新生児を対象とした臨床試験は実施していない。
10. 相互作用
10.2 併用注意
| 網膜症を引き起こすおそれがある薬剤 ヒドロキシクロロキン等 | 併用により視野障害のリスクが増大するおそれがある。 | 共に網膜障害を引き起こす可能性があるため。 |
| 緑内障を引き起こすおそれがある薬剤 プレドニゾロン等 | 併用により視野障害のリスクが増大するおそれがある。 | 共に視野障害を引き起こす可能性があるため。 |
| フェニトイン、ホスフェニトインナトリウム水和物 | 本剤と併用した場合にフェニトインの血中濃度が低下する可能性がある。 | 機序不明 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 視野障害、視力障害(いずれも頻度不明)
11.1.2 視神経萎縮、視神経炎(いずれも頻度不明)
11.1.3 てんかん重積状態(5%未満)、ミオクローヌス発作(頻度不明)
11.1.4 呼吸障害(頻度不明)
呼吸停止、呼吸困難、呼吸不全等の呼吸障害があらわれることがある。
11.1.5 脳症症状(鎮静、昏迷、錯乱、意識障害等)(いずれも頻度不明)[8.3参照]
11.1.6 頭部MRI異常(脳の器質的異常)(頻度不明)
11.2 その他の副作用
| 5%以上 | 5%未満 | 頻度不明 | |
| 精神障害 | 激越、不眠症 | 興奮、攻撃性、神経過敏、うつ病、妄想反応、軽躁、躁病、精神病性障害、自殺企図、幻覚 | |
| 神経系障害 | 傾眠 | 浮動性めまい | 会話障害、頭痛、錯感覚、注意力障害、記憶障害、精神的機能障害(思考障害)、振戦、協調運動異常(運動失調)、運動障害(ジストニア、ジスキネジア、筋緊張亢進) |
| 一般・全身障害および投与部位の状態 | 疲労、浮腫、易刺激性 | ||
| 代謝および栄養状態 | 食欲減退 | ||
| 胃腸障害 | 悪心、嘔吐、腹痛 | ||
| 眼障害 | 霧視、複視、眼振 | ||
| 皮膚および皮下組織障害 | 発疹、血管浮腫、蕁麻疹、脱毛症 | ||
| 筋骨格系および結合組織障害 | 関節痛 | ||
| 血液およびリンパ系障害 | 貧血 | ||
| 臨床検査 | ALT減少 | 体重増加、AST減少 |
12. 臨床検査結果に及ぼす影響
12.1 本剤の投与によりALTについては検査値が30〜100%低下するとの報告があり5)、本剤投与中の患者ではALT及びASTの検査値が影響を受けて低下することがあるので、本剤投与中の患者で肝機能を評価する場合にはALT及びAST以外の肝機能検査項目(LDHなど)も考慮すること。
12.2 本剤は尿中のアミノ酸量を増加させるため、特定のまれな遺伝性代謝疾患(α-アミノアジピン酸尿症など)の検査結果が偽陽性となる可能性がある。
13. 過量投与
13.1 症状
外国における過量投与の報告として、最も多く報告されている症状は傾眠又は昏睡で、その他として回転性めまい、頭痛、精神病、呼吸抑制又は無呼吸、徐脈、低血圧、激越、易刺激性、錯乱、異常行動、会話障害といった症状が報告されている。
13.2 処置
本剤の過量投与時の解毒剤は知られていない。過量投与に対しては未吸収の薬物を排出させる処置を検討すること。活性炭はビガバトリンを大量に吸着できない。また、血液透析による本剤の除去の有効性は不明である。なお、本剤の治療を受けた腎不全患者における個々の症例報告では、血液透析により本剤の血中濃度が40〜60%低下したとの報告がある。
14. 適用上の注意
14.1 薬剤交付時の注意
本剤は必要量に再分包して交付すること。薬剤を交付する際には、服用の直前に適量の水に溶解した後、速やかに全量を服用するよう指導すること。
15. その他の注意
15.1 臨床使用に基づく情報
海外で実施された複数の抗てんかん薬における、てんかん、精神疾患等を対象とした199のプラセボ対照比較試験の検討結果において、自殺念慮及び自殺企図の発現のリスクが、抗てんかん薬の服用群でプラセボ群と比較して約2倍高く(抗てんかん薬服用群:0.43%、プラセボ群:0.24%)、抗てんかん薬の服用群では、プラセボ群と比べ1000人あたり1.9人多いと計算された(95%信頼区間:0.6-3.9)。また、てんかん患者のサブグループでは、プラセボ群と比べ1000人あたり2.4人多いと計算されている。
15.2 非臨床試験に基づく情報
15.2.1 脳への影響
15.2.2 眼毒性
16. 薬物動態
16.1 血中濃度
日本人乳幼児点頭てんかん患者9名にビガバトリン(散剤)37.5〜75mg/kg/回(1日量75〜150mg/kg)を反復経口投与したとき、反復投与12〜16日目における50mg/kg/回の用量で標準化した血漿中ビガバトリン(R,S体)及びビガバトリンS体(活性体)の濃度の推移は下図のとおりであり、薬物動態パラメータは下表のとおりであった18)。
| 薬物動態パラメータ(平均値±標準偏差) | |||
| Ctrough(μg/mL) | C2h(μg/mL) | AUCτ(μg・h/mL) | |
| ビガバトリン(R,S体) | 5.28±1.74 | 60.84±15.44 | 315.83±62.85 |
| ビガバトリンS体 | 3.21±0.99 | 25.73±7.24 | 145.03±30.59 |
日本人乳幼児点頭てんかん患者にビガバトリン(散剤)を反復投与したときの定常状態における平均血漿中ビガバトリン濃度推移(50mg/kg/回の用量で標準化)
16.2 吸収
16.2.1 食事の影響
日本人健康成人6名に本剤(散剤)2gを空腹時単回経口投与又は本剤(散剤)2gを1日1回5日間食後反復経口投与したとき、血漿中未変化体(ビガバトリン)の薬物動態パラメータは下表のとおりであった。空腹時投与と比べ、食後投与においてCmaxの若干の低下がみられたものの、AUCに差はみられなかった19)。
| 投与量 | 測定時期 | Cmax(μg/mL) | Tmax(h) | t1/2(h) | AUC(μg・h/mL)注1) | |
| 空腹時単回投与 | 2.0g | − | 66.7(21.4) | 1.0(46.6) | 7.0(19.4) | 270(20.9) |
| 食後反復投与 | 2.0g 1日1回 | 1日目 | 42.6(12.0) | 1.7(54.3) | 5.6(13.4) | 255(13.8) |
| 5日目 | 42.5(18.9) | 1.7(70.1) | 6.0(37.3) | 291(16.0) |
16.3 分布
16.3.1 蛋白結合
本剤はin vitroにおいてヒト血漿タンパクにほとんど結合しなかった(平衡透析法)20)。
16.4 代謝
16.5 排泄
[16.4参照]
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者(成人)での体内動態
腎機能障害患者(成人)16名(軽度〜中等度[CLcr:40〜79mL/min]
8名、中等度〜重度[CLcr:10〜39mL/min]:8名)及び腎機能正常被験者(成人)8名に、ビガバトリン(液剤)0.75gを単回経口投与したときのラセミ体(R,S体)及びエナンチオマー(S体)の血漿中薬物動態について検討した。
その結果、腎機能障害の程度に伴って、AUCの増加及びt1/2の延長が認められたが、Cmax及びTmaxへの影響はわずかであった(下表)24)(外国人データ)。[9.2参照]
その結果、腎機能障害の程度に伴って、AUCの増加及びt1/2の延長が認められたが、Cmax及びTmaxへの影響はわずかであった(下表)24)(外国人データ)。[9.2参照]
| 用量0.75g | 評価例数 | PKパラメータ | ||||
| 平均値(CV%) | ||||||
| Cmax(μg/mL) | AUCinf(μg・h/mL) | CL/F(L/h) | t1/2(h) | Tmax注2)(h) | ||
| ラセミ体として測定 | ||||||
| 正常 | 8 | 29.5(7.6) | 148.2(14.0) | 5.2(14.6) | 8.1(15.3) | 0.75(0.33-1.00) |
| 軽度〜中等度 | 8 | 29.5(16.7) | 196.2(18.0) | 3.9(17.3) | 12.1(16.6) | 0.75(0.33-1.00) |
| 中等度〜重度 | 8 | 33.8(23.3) | 523.5(38.2) | 1.7(44.6) | 23.4(37.1) | 0.75(0.33-1.00) |
| S体として測定 | ||||||
| 正常 | 8 | 9.5(26.2) | 57.3(24.2) | 6.9(24.3) | 7.7(22.2) | 0.50(0.33-1.00) |
| 軽度〜中等度 | 8 | 10.4(23.0) | 83.0(13.7) | 4.6(14.7) | 9.6(10.9) | 0.75(0.33-1.00) |
| 中等度〜重度 | 8 | 12.7(22.8) | 143.2(21.6) | 2.7(22.6) | 12.4(22.2) | 0.625(0.50-1.00) |
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国内第III相試験
生後4週以上2歳未満の点頭てんかん患者(有効性解析対象13例)を対象に、本剤を50mg/kg/日から投与を開始し、6日〜8週間かけて3g/日を上限として150mg/kg/日まで漸増投与して適切な用量を決定した後(用量調整期)、その用量を2週間継続投与(スパズムの再発・悪化が確認された場合は、1回のみ増量することができるものとし、最高投与量は3g/日を上限に150mg/kg/日、増量の判断は7日目の投与前までに実施)した(維持投与期)。主要評価項目である主要評価スパズム判定日(維持投与期開始前2日間)においてスパズム頻度がベースラインと比較して50%以上減少した患者は、13例中8例(61.5%、95%信頼区間:31.6〜86.1%)であった。また、維持投与期スパズム判定日(維持投与期最終日及びその前日)において点頭てんかんの完全消失(スパズム及び脳波におけるヒプスアリスミアの消失)が認められた患者は13例中4例(30.8%)であった。
副作用は13例中11例(84.6%)に認められた。主な副作用は、傾眠が6例(46.2%)、激越及びアラニンアミノトランスフェラーゼ減少が各4例(30.8%)、不眠症が2例(15.4%)であった。18)。
副作用は13例中11例(84.6%)に認められた。主な副作用は、傾眠が6例(46.2%)、激越及びアラニンアミノトランスフェラーゼ減少が各4例(30.8%)、不眠症が2例(15.4%)であった。18)。
17.1.2 国内長期投与試験
先行する国内第III相試験の維持投与期を終了し、有効性が認められ、安全性に問題がなかった点頭てんかん患者(ケース1、有効性解析対象7例)、本剤により治療中の生後4週以上6歳未満の点頭てんかん患者(ケース2、有効性解析対象2例)、並びに本剤による治療歴がなく、スパズムが認められる2歳以上6歳未満の点頭てんかん患者(ケース3、有効性解析対象5例)を対象とした長期試験において、評価項目であるスパズム頻度がベースラインと比較して50%以上減少した患者、並びに点頭てんかんの完全消失(スパズム及び脳波におけるヒプスアリスミアの消失)が認められた患者は下表のとおりであった。
| 評価項目 | スパズムがベースラインと比較して50%以上減少注1) | 点頭てんかんの完全消失注2) | |||
| 対象集団 | ケース1 | ケース3 | ケース1 | ケース3 | |
| 評価時点 | 先行第III相試験維持投与期スパズム判定日 | 6/7(85.7) | − | 4/7(57.1) | − |
| 本試験開始後16週 | 7/7(100.0) | 3/4(75.0) | 4/7(57.1) | 1/4(25.0) | |
| 本試験開始後32週 | 6/6(100.0) | 3/4(75.0) | 3/6(50.0) | 1/4(25.0) | |
| 本試験開始後56週 | 5/5(100.0) | 3/3(100.0) | 2/5(40.0) | 1/3(33.3) | |
副作用は17例中10例(58.8%)23件に認められた。主な副作用は、激越4例(23.5%)、傾眠2例(11.8%)、不眠症、浮動性めまい、てんかん重積状態、喘息、食欲減退、細気管支炎、副鼻腔炎、アデノウイルス感染、アラニンアミノトランスフェラーゼ減少、臨床検査異常及び核磁気共鳴画像異常の各1例(5.9%)であった25)。
17.1.3 海外第III相単盲検試験
3ヵ月以内に点頭てんかんと診断された2歳未満の患者(有効性解析対象221例)を対象に、本剤(フィルムコート錠)低用量(18〜36mg/kg/日)又は高用量(100〜148mg/kg/日)を1日2回14日間経口投与した。
主要評価項目である本剤投与開始後14日以内のいずれかの時点から連続7日間のスパズム及びヒプスアリスミアの消失注3)が認められた患者は、低用量群で114例中8例(7.0%)、高用量群で107例中17例(15.9%)であり、低用量群に比べて高用量群で有意に高かった(p=0.0375、カイ二乗検定)。
安全性解析対象222例に218件の副作用が認められた。主な副作用は、鎮静、傾眠、易刺激性、不眠症及び睡眠障害であった26)。
主要評価項目である本剤投与開始後14日以内のいずれかの時点から連続7日間のスパズム及びヒプスアリスミアの消失注3)が認められた患者は、低用量群で114例中8例(7.0%)、高用量群で107例中17例(15.9%)であり、低用量群に比べて高用量群で有意に高かった(p=0.0375、カイ二乗検定)。
安全性解析対象222例に218件の副作用が認められた。主な副作用は、鎮静、傾眠、易刺激性、不眠症及び睡眠障害であった26)。
17.1.4 海外第III相プラセボ対照二重盲検試験
新たに点頭てんかんと診断された生後1〜18ヵ月の患者(有効性解析対象40例)を対象に、プラセボ又は本剤(散剤)50mg/kg/日の1日2回経口投与から開始し、スパズムの消失を認めない場合は150mg/kg/日まで増量し、投与開始後5日目まで投与した(二重盲検期)。
主要評価項目である二重盲検期の最終2日間(各日2時間)でのベースラインからのスパズム頻度減少率(%)(最小二乗平均値[95%信頼区間])は、プラセボ群(20例)40.5[−17,70]、本剤群(20例)54.4[12,76]であり、プラセボ群と本剤群の間に統計学的な有意差は認められなかった(p=0.562、対数変換したスパズム頻度減少率に対する投与群、施設を因子、対数変換したベースラインのスパズム頻度を共変量とした共分散分析)。一方、副次評価項目である二重盲検期の最終2日間(各日24時間)でのベースラインからのスパズム頻度減少率(%)(最小二乗平均値[95%信頼区間])は、プラセボ群17.0[−59,57]、本剤群68.9[42,83]であり、群間に統計学的有意差が認められた(p=0.030)。
二重盲検期では、ビガバトリン群20例中12例(60.0%)及びプラセボ群20例中6例(30.0%)に1件以上の有害事象が認められた。ビガバトリン群における主な有害事象は傾眠状態(8例)であった27)。
主要評価項目である二重盲検期の最終2日間(各日2時間)でのベースラインからのスパズム頻度減少率(%)(最小二乗平均値[95%信頼区間])は、プラセボ群(20例)40.5[−17,70]、本剤群(20例)54.4[12,76]であり、プラセボ群と本剤群の間に統計学的な有意差は認められなかった(p=0.562、対数変換したスパズム頻度減少率に対する投与群、施設を因子、対数変換したベースラインのスパズム頻度を共変量とした共分散分析)。一方、副次評価項目である二重盲検期の最終2日間(各日24時間)でのベースラインからのスパズム頻度減少率(%)(最小二乗平均値[95%信頼区間])は、プラセボ群17.0[−59,57]、本剤群68.9[42,83]であり、群間に統計学的有意差が認められた(p=0.030)。
二重盲検期では、ビガバトリン群20例中12例(60.0%)及びプラセボ群20例中6例(30.0%)に1件以上の有害事象が認められた。ビガバトリン群における主な有害事象は傾眠状態(8例)であった27)。
17.1.5 海外第III相クロスオーバー比較試験(他剤比較)
結節性硬化症による点頭てんかんと新たに診断された生後1ヵ月〜2歳の患者(有効性解析対象22例)を対象に、本剤(錠剤)150mg/kg/日を1日2回又はヒドロコルチゾン15mg/kg/日を1日1回1ヵ月間経口投与した後、スパズムの消失を認めなかった場合及び/又は忍容性に問題があった場合にはもう一方の群に移行して1ヵ月間経口投与した。
主要評価項目である投与1ヵ月目におけるスパズムの消失が認められた患者は、本剤群で11例中11例(100%)、ヒドロコルチゾン群で11例中4例(36.4%)であり、ヒドロコルチゾン群と比較して本剤群で有意に高かった(p=0.001、イェーツ補正したカイ二乗検定)。また、ヒドロコルチゾン群から本剤群に移行した7例では投与2ヵ月目に全例でスパズムの消失が認められた。なお、本剤群からヒドロコルチゾン群に移行した患者はいなかった。
少なくとも1件発現した有害事象は、ビガバトリン群で18例中5例(1次治療3例、2次治療2例)及びヒドロコルチゾン群で11例中9例であった。主な副作用は、ビガバトリン群で傾眠状態及び運動過多(障害)であり、ヒドロコルチゾン群で興奮性亢進、睡眠障害及び体重増加であった28)。
主要評価項目である投与1ヵ月目におけるスパズムの消失が認められた患者は、本剤群で11例中11例(100%)、ヒドロコルチゾン群で11例中4例(36.4%)であり、ヒドロコルチゾン群と比較して本剤群で有意に高かった(p=0.001、イェーツ補正したカイ二乗検定)。また、ヒドロコルチゾン群から本剤群に移行した7例では投与2ヵ月目に全例でスパズムの消失が認められた。なお、本剤群からヒドロコルチゾン群に移行した患者はいなかった。
少なくとも1件発現した有害事象は、ビガバトリン群で18例中5例(1次治療3例、2次治療2例)及びヒドロコルチゾン群で11例中9例であった。主な副作用は、ビガバトリン群で傾眠状態及び運動過多(障害)であり、ヒドロコルチゾン群で興奮性亢進、睡眠障害及び体重増加であった28)。
注3)保護者の観察に基づき、スパズムが連続7日間消失していることが確認され、また、消失7日目から3日以内に、1回以上の睡眠−覚醒−睡眠サイクルを含む8時間の閉鎖回路ビデオ脳波モニタリング(CCTV EEG)により、スパズム及びヒプスアリスミアが認められないことが確認された患者と定義した。
18. 薬効薬理
18.1 作用機序
18.2 抗けいれん作用
19. 有効成分に関する理化学的知見
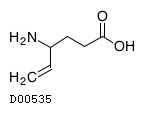
19.1. ビガバトリン
| 一般的名称 | ビガバトリン |
|---|---|
| 一般的名称(欧名) | Vigabatrin |
| 化学名 | (±)-4-Amino-5-hexenoic acid |
| 分子式 | C6H11NO2 |
| 分子量 | 129.16 |
| 融点 | 171〜177℃ |
| 物理化学的性状 | 本品は白色の粉末である。 本品は水に溶けやすく、メタノール又はエタノール(95)に溶けにくい。 |
| KEGG DRUG |
|
21. 承認条件
21.1 医薬品リスク管理計画を策定の上、適切に実施すること。
21.2 本剤による視野障害、視力障害等の重篤な有害事象に対して、他の医療機関との連携も含めて十分に対応できる体制が確認できた医療機関において、点頭てんかんの診断、治療に精通し、本剤の適正使用について十分に理解している医師によって本剤の処方が行われ、本剤の適正使用について十分に理解している眼科医により定期的な診察及び検査が実施されるとともに、本剤の適正使用について十分に理解している薬剤師によって調剤が行われるよう、製造販売にあたって本剤に関する管理者の設置も含め必要な措置を講じること。
21.3 本剤の投与が適切と判断される患者を対象に、あらかじめ患者又は代諾者に安全性及び有効性が文書によって説明され、文書による同意を得てから本剤の投与が開始されるよう、厳格かつ適正な措置を講じること。
21.4 国内での治験症例が極めて限られていることから、製造販売後、一定数の症例に係るデータが集積されるまでの間は、全症例を対象とした使用成績調査を実施することにより、本剤使用患者の患者背景を把握するとともに、本剤の安全性及び有効性に関するデータを早期に収集し、本剤の適正使用に必要な措置を講じること。
22. 包装
60包
23. 主要文献
- Horton M,et al., J Child Neurol., 24 (5), 1543-6, (2009)
- 社内資料:ラットの胚・胎児発生に関する試験(2016年3月28日承認、CTD2.6.6.6)
- 社内資料:ウサギの胚・胎児発生に関する試験(2016年3月28日承認、CTD2.6.6.6)
- 社内資料:ラットの出生前及び出生後の発生並びに母体の機能に関する試験(2016年3月28日承認、CTD2.6.6.6)
- Williams A,et al., Aust N Z J Med., 24 (1), 65, (1994) »PubMed
- 社内資料:マウスがん原性試験(2016年3月28日承認、CTD2.6.6.5)
- 社内資料:ラット1年間経口投与毒性試験(回復試験含む)(2016年3月28日承認、CTD2.6.6.3)
- 社内資料:イヌ1年間経口投与毒性試験(2016年3月28日承認、CTD2.6.6.3)
- 社内資料:サル6年間経口投与毒性試験(2016年3月28日承認、CTD2.6.6.3)
- 社内資料:イヌ1年間経口投与毒性試験の回復試験(2016年3月28日承認、CTD2.6.6.3)
- Walzer M,et al., Neurotoxicology., 32 (6), 963-74, (2011) »PubMed
- Rasmussen A D,et al., Neurotoxicology., 46, 137-44, (2015) »PubMed
- Bottomley A L,et al., Toxicol Pathol., 43 (7), 1015-24, (2015) »PubMed
- 社内資料:ラットの眼毒性試験(2016年3月28日承認、CTD2.6.6.8)
- 社内資料:アルビノラットと有色ラットの網膜への影響の比較(2016年3月28日承認、CTD2.6.6.8)
- 社内資料:有色ラットによる眼毒性試験(2016年3月28日承認、CTD2.6.6.8)
- Izumi Y,et al., Epilepsia., 45 (9), 1043-8, (2004) »PubMed
- 社内資料:点頭てんかんを対象とした第3相試験(2016年3月28日承認、CTD2.7.6.2)
- 社内資料:日本人健康成人被験者におけるビガバトリン単回及び反復投与時の安全性及び薬物動態(2016年3月28日承認、CTD2.7.2.2)
- 社内資料:In vitroにおけるビガバトリンの血漿タンパク結合(2016年3月28日承認、CTD2.7.2.2)
- 社内資料:健康被験者に14C-ビガバトリンを単回経口投与したときの薬物動態及び代謝(2016年3月28日承認、CTD2.7.2.2)
- 社内資料:In vitroにおけるビガバトリンの酵素誘導(CYP2B6,2C9,2C19,3A4)(2016年3月28日承認、CTD2.7.2.2)
- 社内資料:In vitroにおけるビガバトリンの酵素誘導(CYP1A2,3A4/5)(2016年3月28日承認、CTD2.7.2.2)
- 社内資料:外国人腎機能障害患者における薬物動態(2016年3月28日承認、CTD2.7.2.2)
- 社内資料:点頭てんかんを対象とした長期投与試験(2016年3月28日承認、CTD2.7.6.2)
- 社内資料:海外第III相単盲検試験(2016年3月28日承認、CTD2.7.6.2)
- 社内資料:海外第III相プラセボ対照二重盲検試験(2016年3月28日承認、CTD2.7.6.2)
- 社内資料:海外第III相クロスオーバー比較試験(他剤比較)(2016年3月28日承認、CTD2.7.6.2)
- 社内資料:In vitroにおけるビガバトリンのGABA-T阻害作用(2016年3月28日承認、CTD2.6.2.2)
- Jung M J,et al., J Neurochem., 29 (5), 797-802, (1977) »PubMed
- Bohlen P,et al., Brain Res., 167 (2), 297-305, (1979) »PubMed
- Iadarola M J,et al., Brain Res Bull., 5, 13-9, (1980)
- Kubova H,et al., Epilepsia., 51 (3), 469-72, (2010) »PubMed
- 社内資料:マウスにおける薬物誘発痙攣に対するビガバトリン単回経口投与の作用(2016年3月28日承認、CTD2.6.2.2)
- 社内資料:ビガバトリンの抗痙攣作用(2016年3月28日承認、CTD2.6.2.2)
- Shin C,et al., Brain Res., 398 (2), 370-4, (1986) »PubMed
- Schechter P J,et al., Eur J Pharmacol., 45 (5), 319-28, (1977)
24. 文献請求先及び問い合わせ先
文献請求先
アルフレッサファーマ株式会社
製品情報部
〒540-8575
大阪市中央区石町二丁目2番9号
電話:06-6941-0306
FAX:06-6943-8212
製品情報問い合わせ先
アルフレッサファーマ株式会社
製品情報部
〒540-8575
大阪市中央区石町二丁目2番9号
電話:06-6941-0306
FAX:06-6943-8212
26. 製造販売業者等
26.1 製造販売元
サノフィ株式会社
〒163-1488
東京都新宿区西新宿三丁目20番2号
26.2 販売元
アルフレッサファーマ株式会社
大阪市中央区石町二丁目2番9号