医薬品情報
| 総称名 | ベルソムラ |
|---|---|
| 一般名 | スボレキサント |
| 欧文一般名 | Suvorexant |
| 製剤名 | スボレキサント錠 |
| 薬効分類名 | オレキシン受容体拮抗薬 不眠症治療薬 |
| 薬効分類番号 | 1190 |
| ATCコード | N05CJ01 |
| KEGG DRUG |
D10082
スボレキサント
|
| KEGG DGROUP |
DG03202
睡眠薬
|
| JAPIC | 添付文書(PDF) |
添付文書情報2026年3月 改訂(第5版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ベルソムラ錠10mg | Belsomra Tablets 10mg | MSD | 1190023F3027 | 69.3円/錠 | 習慣性医薬品, 処方箋医薬品 |
| ベルソムラ錠15mg | Belsomra Tablets 15mg | MSD | 1190023F1024 | 90.8円/錠 | 習慣性医薬品, 処方箋医薬品 |
| ベルソムラ錠20mg | Belsomra Tablets 20mg | MSD | 1190023F2020 | 109.9円/錠 | 習慣性医薬品, 処方箋医薬品 |
2. 禁忌
次の患者には投与しないこと
4. 効能または効果
5. 効能または効果に関連する注意
二次性不眠症に対する本剤の有効性及び安全性は確立されていない。
6. 用法及び用量
通常、成人にはスボレキサントとして1日1回20mgを、高齢者には1日1回15mgを就寝直前に経口投与する。
7. 用法及び用量に関連する注意
7.2 入眠効果の発現が遅れるおそれがあるため、本剤の食事と同時又は食直後の服用は避けること。食後投与では、空腹時投与に比べ、投与直後のスボレキサントの血漿中濃度が低下することがある。[16.2.1参照]
7.3 他の不眠症治療薬と併用したときの有効性及び安全性は確立されていない。
8. 重要な基本的注意
8.1 本剤の影響が服用の翌朝以後に及び、眠気、注意力・集中力・反射運動能力等の低下が起こることがあるので、自動車の運転など危険を伴う機械の操作に従事させないように注意すること。[17.3.1参照]
8.2 症状が改善した場合は、本剤の投与継続の要否について検討し、本剤を漫然と投与しないよう注意すること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 ナルコレプシー又はカタプレキシーのある患者
症状を悪化させるおそれがある。
9.1.2 重度の呼吸機能障害を有する患者
重度の呼吸機能障害を有する患者を対象とした臨床試験は実施していない。[17.3.1参照]
9.1.3 脳に器質的障害のある患者
作用が強くあらわれるおそれがある。
9.3 肝機能障害患者
9.3.1 重度の肝機能障害のある患者
スボレキサントの血漿中濃度を上昇させるおそれがある。[16.6.3参照]
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。動物実験(ラット)では、交配前、交配期間中及び妊娠初期に臨床曝露量の70倍を投与した場合、黄体数、着床数及び生存胎児数の減少が、妊娠期に臨床曝露量の86倍を投与した場合、胎児体重の減少が認められた。また、妊娠から授乳期に臨床曝露量の49倍を投与した場合、出生児に一過性の体重低値が認められた。
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。外国人女性12例を対象とした乳汁移行試験成績から、スボレキサント及びその水酸化代謝物が母乳中へ移行することが認められている。本剤20mgを経口投与した時の相対的乳児投与量(RID)は母体投与量の1%未満であり、水酸化代謝物の移行はスボレキサントより低かった。母乳中のスボレキサントに対する水酸化代謝物の曝露量比は約0.13であった。動物実験(ラット)でスボレキサントが乳汁中へ移行することが報告されている。
9.7 小児等
小児等を対象とした臨床試験は実施していない。
9.8 高齢者
患者の状態を観察しながら投与すること。一般に高齢者では生理機能が低下している。高齢者での薬物動態試験において、非高齢者と比較して血漿中濃度が高くなる傾向が認められている。[16.6.1参照]
10. 相互作用
相互作用序文
スボレキサントは主に薬物代謝酵素CYP3Aによって代謝される。また、弱いP糖蛋白(腸管)への阻害作用を有する。[16.4参照]
薬物代謝酵素用語
CYP3A
薬物代謝酵素用語
P糖蛋白
10.1 併用禁忌
10.2 併用注意
| アルコール(飲酒) [16.7.1参照] | 精神運動機能の相加的な低下を生じる可能性がある。本剤を服用時に飲酒は避けさせること。 | 本剤及びアルコールは中枢神経系に対する抑制作用を有するため、相互に作用を増強させるおそれがある。 |
| 中枢神経抑制剤(フェノチアジン誘導体、バルビツール酸誘導体等) | 中枢神経系に対する抑制作用を増強させるおそれがある。 | 本剤及びこれらの薬剤は中枢神経系に対する抑制作用を有するため、相互に作用を増強させるおそれがある。 |
| CYP3Aを中等度に阻害する薬剤(ジルチアゼム、ベラパミル、フルコナゾール等) [7.4、16.7.2参照] | 傾眠、疲労等の本剤の副作用が増強するおそれがあるため、併用する際には1日1回10mgへの減量を考慮するとともに、患者の状態を慎重に観察すること。 | スボレキサントの代謝酵素であるCYP3Aを中等度に阻害し、スボレキサントの血漿中濃度を上昇させる。 |
| CYP3Aを強く誘導する薬剤(リファンピシン、カルバマゼピン、フェニトイン等) [16.7.2参照] | 本剤の作用を減弱させるおそれがある。 | スボレキサントの代謝酵素であるCYP3Aを強く誘導し、スボレキサントの血漿中濃度を低下させる。 |
| ジゴキシン [16.7.3参照] | ジゴキシンの血漿中濃度を上昇させるおそれがある。本剤と併用する場合は、ジゴキシンの血漿中濃度をモニタリングすること。 | スボレキサントはP糖蛋白阻害作用を有する。 |
11. 副作用
11.2 その他の副作用
| 1〜5%未満 | 1%未満 | 頻度不明 | |
| 心臓障害 | 動悸 | ||
| 胃腸障害 | 悪心、嘔吐 | ||
| 一般・全身障害及び投与部位の状態 | 疲労 | ||
| 神経系障害 | 傾眠、頭痛、浮動性めまい | 睡眠時麻痺 | |
| 精神障害 | 悪夢 | 異常な夢、入眠時幻覚 | 睡眠時随伴症注)、夢遊症注)、傾眠時幻覚注)、不安、激越 |
| 皮膚及び皮下組織障害 | そう痒症 |
13. 過量投与
13.1 徴候、症状
本剤の過量投与に関する情報は少ない。外国人健康成人に本剤120〜240mgを朝投与した臨床試験で、用量依存的に傾眠の発現率及び持続時間が増加し、脈拍数が一過性に低下する傾向がみられた。外国人健康成人に本剤240mgを朝投与した臨床試験では、胸痛及び呼吸抑制が報告された。
13.2 処置
血液透析は本剤の除去に有用かどうかは不明である。スボレキサントは蛋白質結合能が高いため、血液透析では除去されないと考えられる。多剤服用の可能性を考慮する。
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔を起こして縦隔洞炎等の重篤な合併症を併発することがある。
15. その他の注意
15.2 非臨床試験に基づく情報
15.2.1 ラットの2年間がん原性試験では、臨床曝露量の36倍の投与により肝細胞腺腫及び臨床曝露量の11倍の投与により甲状腺濾胞細胞腺腫の発現頻度が増加したが、これらの変化はげっ歯類に特異的な肝酵素誘導及び甲状腺ホルモン産生増加の二次的な変化と考えられた。一方、rasH2トランスジェニックマウスでは、臨床曝露量の105倍までの用量を6ヵ月間経口投与しても、がん原性を示唆する変化は認められなかった。
15.2.2 ラットの2年間がん原性試験において、臨床曝露量の11倍(雄)及び18倍(雌)以上の用量で網膜萎縮の発現頻度が増加した。薬効量を大きく超えた用量のオレキシン受容体拮抗薬をラットに投与すると明期における覚醒時間が増加したこと、スボレキサントを投与した有色ラットの網膜萎縮の発現はアルビノラットよりも遅く、その発現率及び重症度も低かったことが報告されている。さらに、イヌに臨床曝露量の84倍を9ヵ月間投与しても網膜変化はみられていない。これらのことからラットがん原性試験でみられた網膜萎縮は、アルビノラットで自然発生的に生じることが知られている加齢及び光誘発性の網膜萎縮の発現頻度が、スボレキサントの薬理作用を介した網膜への光照射の増加により増加したことを反映した、ラット特有の変化と考えられた。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
日本人健康成人(12例)に、本剤40mgを空腹時単回経口投与した後のスボレキサントは速やかに吸収され、投与後1.5時間(1.0〜3.0時間)で最高血漿中濃度(Cmax)に達し、平均半減期(t1/2)は10.0時間であった(表1)。
| AUC0-∞†(μM・hr) | Cmax†(μM) | Tmax††(hr) | t1/2§(hr) |
| 12.15(10.97,13.46) | 1.007(0.858,1.182) | 1.5(1.0,3.0) | 10.0±1.0 |
16.1.2 単回投与
健康成人(16例)に、本剤10〜40mgを空腹時単回経口投与したところ、スボレキサントの曝露量は用量比例性を下回った(外国人データ)(表2)。
| AUC0-∞†(μM・hr) | Cmax†(μM) | Tmax††(hr) | t1/2§(hr) | |
| 10mg | 5.32(4.55,6.23) | 0.456(0.403,0.516) | 1.5(1.0,4.0) | 12.1±1.8 |
| 20mg | 9.51(8.12,11.14) | 0.646(0.572,0.731) | 1.0(1.0,4.0) | 12.5±2.6 |
| 40mg | 16.21(13.85,18.98) | 0.956(0.845,1.082) | 2.0(1.0,4.0) | 12.6±2.5 |
16.1.3 反復投与
健康成人(30例)に、本剤10〜100mgを1日1回14日間反復投与したとき、3日目までに定常状態に到達し、スボレキサント40mgの平均t1/2(約12時間、95%信頼区間:12.0〜13.1時間)から予想される値と一致した。AUC0-24hrの累積係数は1.21〜1.60で、いずれの用量でも類似していた(外国人データ)。
16.2 吸収
本剤20mgを投与した際の平均絶対バイオアベイラビリティは62%(5〜95パーセンタイル:55〜69%)であると推定された。
16.2.1 食事の影響
本剤40mgを低脂肪食摂取後に単回経口投与した場合、空腹時と比較してスボレキサントのCmaxは23%増加したが、AUCは変化しなかった。Tmaxは1時間延長した(日本人データ)。
本剤40mgを高脂肪食摂取後に単回経口投与した場合、空腹時と比較してスボレキサントのCmaxは9%増加したが、AUCは変化しなかった。Tmaxは1.5時間延長した(外国人データ)。[7.2参照]
本剤40mgを高脂肪食摂取後に単回経口投与した場合、空腹時と比較してスボレキサントのCmaxは9%増加したが、AUCは変化しなかった。Tmaxは1.5時間延長した(外国人データ)。[7.2参照]
16.3 分布
スボレキサントの平均分布容積は約49Lであった。
スボレキサントのヒト血漿蛋白結合率は高く(>99%)、赤血球に特異的に分布することはなかった。スボレキサントは、ヒト血清アルブミン及びα1-酸性糖蛋白質のいずれにも結合した。
スボレキサントのヒト血漿蛋白結合率は高く(>99%)、赤血球に特異的に分布することはなかった。スボレキサントは、ヒト血清アルブミン及びα1-酸性糖蛋白質のいずれにも結合した。
16.4 代謝
スボレキサントは主として代謝により消失し、その代謝には主にCYP3Aが関与し、CYP2C19もわずかに関与していた。血漿中には主にスボレキサント及びその水酸化代謝物が認められた。この代謝物は脳内で薬理作用を示さないと考えられる。
16.5 排泄
スボレキサントの主な排泄経路は糞便を介するものであり、経口投与した14C標識スボレキサントの約66%が糞便中に排泄されるのに対し、尿中への排泄は23%であった。スボレキサントは主として代謝物として排泄され、糞便中及び尿中のスボレキサントは投与量の1%未満であった。
16.6 特定の背景を有する患者
16.6.1 年齢
健康成人に、本剤40mgを1日1回14日間反復投与したとき、定常状態でのスボレキサントのAUC0-24hr、Cmax及びt1/2の平均値は、それぞれ、10.64μM・hr、1.080μM及び9.4時間であった。健康高齢者に、本剤40mgを1日1回7日間反復投与したとき、定常状態でのスボレキサントのAUC0-24hr、Cmax及びt1/2の平均値は、それぞれ、17.88μM・hr、1.336μM及び18.4時間であり、健康成人と比べて、AUC0-24hr及びCmaxは高値を示し、t1/2の延長がみられた。高齢不眠症患者及び非高齢不眠症患者に、本剤15mg及び20mgをそれぞれ1日1回反復投与した際の定常状態でのスボレキサントの投与後9時間の血漿中濃度(C9hr)は、それぞれ0.362μM及び0.321μMで同程度であった(外国人データ)。[9.8参照]
16.6.2 腎機能障害
重度腎機能障害患者(CLcr:30mL/min/1.73m2以下)に本剤20mgを単回投与した後のスボレキサントのCmax及びAUCは、健康成人と比較して15%及び22%高かった(外国人データ)。
16.6.3 肝機能障害
中等度肝機能障害患者(Child-Pughスコア7〜9)に本剤20mgを単回投与した後のスボレキサントのCmaxは、健康成人と比較して6%低く、AUCは3%高かった。重度肝機能障害患者(Child-Pughスコア10〜15)での薬物動態は検討していない(外国人データ)。[9.3.1参照]
16.6.4 BMI
本剤20mgを不眠症患者に反復投与した際の定常状態でのスボレキサントのC9hrは、低体重患者(BMI:18.5kg/m2未満、6例)で0.171μM、標準BMI(18.5kg/m2以上、25kg/m2未満、139例)の患者で0.323μM、前肥満患者(BMI:25kg/m2以上、30kg/m2未満、94例)で0.384μM及び肥満患者(BMI:30kg/m2以上、42例)で0.353μMであった(外国人データ)。
16.7 薬物相互作用
16.7.1 アルコール
16.7.2 スボレキサントの薬物動態に対する併用薬の影響
(1)ケトコナゾール
(2)ジルチアゼム
(3)リファンピシン
本剤(40mg単回)とリファンピシン(600mg 1日1回反復)を併用した際、スボレキサントのCmax及びAUCは64%及び88%減少した(外国人データ)。[10.2参照]
16.7.3 併用薬の薬物動態に対するスボレキサントの影響
(1)In vitro代謝試験
スボレキサントはCYP3A及び腸管のP糖蛋白を阻害する可能性があることが示されている。他のヒトCYP分子種(CYP1A2、CYP2B6、CYP2C8、CYP2C9、CYP2C19、CYP2D6)及びトランスポーター(OATP1B1、BCRP、OCT2)に対しては、臨床的に意味のある阻害を生じる可能性は低いと考えられる。スボレキサントを反復投与することによって、主にCYP分子種によって代謝される薬剤の代謝を誘導する可能性は低い。
(2)ジゴキシン(P糖蛋白基質)
本剤(40mg 1日1回反復)とジゴキシン(0.5mg単回)を併用した際、ジゴキシンのCmax及びAUCは21%及び27%増加した。スボレキサント投与時のジゴキシン濃度は最初の6時間以内に増加した(外国人データ)。[10.2参照]
注)本剤の承認用量は成人には1日20mg、高齢者には1日15mgである。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国際共同第III相臨床試験
プラセボ対照試験において、日本人155例を含む原発性不眠症患者638例[成人(20〜64歳)370例、高齢者(65歳以上)268例]に本剤(成人:20mg、高齢者:15mg)又はプラセボを3ヵ月間投与したとき、患者日誌を用いた主観的評価及びポリソムノグラフィを用いた客観的評価により入眠効果及び睡眠維持効果を評価した結果はそれぞれ表1及び表2のとおりであった2)。
| 評価項目 | 評価時点 | 投与群 | 評価例数 | ベースライン | 変化量 | プラセボとの差† |
| sTSOm | 第1週 | プラセボ群 | 376 | 67.2±40.7 | −10.1±33.9 | −5.6(−10.2,−1.1) |
| 本剤群 | 248 | 63.6±37.3 | −14.5±28.4 | |||
| 1ヵ月時 | プラセボ群 | 365 | 65.7±39.4 | −12.8±41.2 | −5.4(−10.9,0.0) | |
| 本剤群 | 244 | 62.7±36.7 | −16.4±31.5 | |||
| 3ヵ月時 | プラセボ群 | 339 | 66.6±39.9 | −18.9±39.3 | −5.2(−10.2,−0.3) | |
| 本剤群 | 228 | 60.5±34.7 | −20.4±27.5 | |||
| LPS | 第1日夜 | プラセボ群 | 290 | 66.2±44.1 | −21.6±45.2 | −9.6(−14.9,−4.3) |
| 本剤群 | 193 | 68.9±49.7 | −33.4±48.0 | |||
| 1ヵ月時 | プラセボ群 | 272 | 66.2±44.0 | −24.4±51.4 | −10.3(−16.0,−4.6) | |
| 本剤群 | 185 | 67.7±46.7 | −36.0±45.5 | |||
| 3ヵ月時 | プラセボ群 | 251 | 65.7±43.9 | −27.1±52.0 | −8.1(−13.8,−2.3) | |
| 本剤群 | 172 | 65.5±43.7 | −35.2±42.4 |
| 評価項目 | 評価時点 | 投与群 | 評価例数 | ベースライン | 変化量 | プラセボとの差† |
| sTSTm | 第1週 | プラセボ群 | 376 | 315.2±65.2 | 15.3±42.9 | 13.6(6.9,20.3) |
| 本剤群 | 248 | 322.4±57.3 | 27.2±40.8 | |||
| 1ヵ月時 | プラセボ群 | 365 | 317.7±65.3 | 23.4±52.0 | 16.3(7.9,24.8) | |
| 本剤群 | 244 | 322.7±57.7 | 38.7±50.5 | |||
| 3ヵ月時 | プラセボ群 | 339 | 316.7±64.5 | 42.1±56.4 | 10.7(1.9,19.5) | |
| 本剤群 | 228 | 325.4±56.7 | 50.3±55.2 | |||
| WASO | 第1日夜 | プラセボ群 | 287 | 115.1±45.9 | −19.1±47.5 | −32.5(−39.3,−25.7) |
| 本剤群 | 192 | 119.5±46.4 | −54.3±44.7 | |||
| 1ヵ月時 | プラセボ群 | 272 | 113.6±45.0 | −17.9±55.3 | −26.4(−34.3,−18.4) | |
| 本剤群 | 185 | 119.1±46.0 | −47.0±45.4 | |||
| 3ヵ月時 | プラセボ群 | 251 | 115.3±46.0 | −25.3±50.7 | −16.6(−24.8,−8.3) | |
| 本剤群 | 172 | 118.2±46.7 | −42.7±50.5 |
この試験の6ヵ月間における副作用は20.9%(53/254例)に認められ、主な副作用は傾眠4.7%、頭痛3.9%、疲労2.4%であった。[7.1参照]
17.3 その他
17.3.1 臨床薬理試験(外国人データ)
(1)自動車運転能力に対する影響
(2)呼吸機能への影響
(3)記憶、精神運動機能及び平衡機能に対する影響
(4)薬物乱用に対する影響
娯楽目的の多剤使用経験のある健康成人36例に本剤40〜150mgを単回投与したとき、本剤の薬物嗜好性はプラセボより高く、ゾルピデム15及び30mgと同程度であった。
注)本剤の承認用量は成人には1日20mg、高齢者には1日15mgである。
18. 薬効薬理
18.1 作用機序
スボレキサントはオレキシン受容体に選択性が高く可逆的な拮抗薬で、ヒトオレキシン1(OX1)受容体及びオレキシン2(OX2)受容体に対する親和性(Ki値)はそれぞれ0.55及び0.35nMであった6)。
スボレキサントは、覚醒を促進する神経ペプチドであるオレキシンA及びBのOX1及びOX2受容体への結合を可逆的に阻害することにより、脳を覚醒状態から睡眠状態へ移行させ、睡眠を誘発すると考えられる。
スボレキサントはγ-アミノ酪酸(GABA)、セロトニン、ドパミン、ノルアドレナリン、メラトニン、ヒスタミン、アセチルコリン及びオピオイド受容体に対して親和性を示さなかった(Ki>10μM)。
18.2 睡眠に対する作用
スボレキサントを通常の活動期にあるラット(10、30及び100mg/kg)、イヌ(1及び3mg/kg)、サル(10mg/kg)に経口投与すると、覚醒時間が減少し、ノンレム睡眠及びレム睡眠時間が増加した6)。
19. 有効成分に関する理化学的知見
19.1. スボレキサント
| 一般的名称 | スボレキサント |
|---|---|
| 一般的名称(欧名) | Suvorexant |
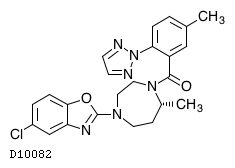
| 化学名 | [(7R)-4-(5-Chloro-1,3-benzoxazol-2-yl)-7-methyl-1,4-diazepan-1-yl][5-methyl-2-(2H-1,2,3-triazol-2-yl)phenyl]methanone |
| 分子式 | C23H23ClN6O2 |
| 分子量 | 450.92 |
| 物理化学的性状 | 白色の粉末である。 メタノールにやや溶けやすく、ヘプタンに極めて溶けにくく、水にほとんど溶けない。 |
| KEGG DRUG |
|
20. 取扱い上の注意
光及び湿気を避けるため、PTPシートのまま保存し、服用直前にPTPシートから取り出すこと。
22. 包装
<ベルソムラ錠10mg>
100錠[10錠(PTP)×10]
<ベルソムラ錠15mg>
100錠[10錠(PTP)×10]
500錠[10錠(PTP)×50]
<ベルソムラ錠20mg>
100錠[10錠(PTP)×10]
500錠[10錠(PTP)×50]
23. 主要文献
- Sun H,et al., J Psychopharmacol., 29, 1159-69, (2015) »PubMed
- Herring WJ,et al., Biol Psychiatry., 79, 136-48, (2016) »PubMed
- Vermeeren A,et al., Sleep., 38, 1803-13, (2015) »PubMed
- Sun H,et al., Respir Med., 109, 416-26, (2015) »PubMed
- Sun H,et al., J Clin Sleep Med., 12, 9-17, (2016) »PubMed
- Winrow CJ,et al., J Neurogenetics., 25, 52-61, (2011)
24. 文献請求先及び問い合わせ先
文献請求先
第一三共株式会社
製品情報センター
〒103-8426
東京都中央区日本橋本町3-5-1
電話:0120-189-132
製品情報問い合わせ先
第一三共株式会社
製品情報センター
〒103-8426
東京都中央区日本橋本町3-5-1
電話:0120-189-132
26. 製造販売業者等
26.1 製造販売元
MSD株式会社
東京都千代田区九段北1-13-12
26.2 販売元
第一三共株式会社
東京都中央区日本橋本町3-5-1