医薬品情報
| 総称名 | モゾビル |
|---|---|
| 一般名 | プレリキサホル |
| 欧文一般名 | Plerixafor |
| 製剤名 | プレリキサホル製剤 |
| 薬効分類名 | CXCR4ケモカイン受容体拮抗剤 |
| 薬効分類番号 | 3399 |
| ATCコード | L03AX16 |
| KEGG DRUG |
D08971
プレリキサホル
|
| JAPIC | 添付文書(PDF) |
添付文書情報2024年5月 改訂(第3版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| モゾビル皮下注24mg | MOZOBIL S.C.Injection | サノフィ | 3399413A1021 | 592719円/瓶 | 劇薬, 処方箋医薬品 |
2. 禁忌
次の患者には投与しないこと
2.1 本剤の成分に対し過敏症の既往歴のある患者
2.2 妊婦又は妊娠している可能性のある女性[9.5参照]
4. 効能または効果
自家末梢血幹細胞移植のための造血幹細胞の末梢血中への動員促進
5. 効能または効果に関連する注意
「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、適応患者の選択を行うこと。
6. 用法及び用量
G-CSF製剤との併用において、通常、成人にはプレリキサホルとして0.24mg/kgを1日1回、末梢血幹細胞採取終了時まで連日皮下投与する。
7. 用法及び用量に関連する注意
7.1 本剤の投与は、G-CSF製剤の投与開始4日目以降、各末梢血幹細胞採取実施9〜12時間前に行う。
7.2 本剤の投与期間は4日間までを目安とすること。
8. 重要な基本的注意
8.1 本剤は、造血幹細胞移植について十分な知識・経験を持つ医師のもとで、本剤の投与が適切と判断される患者についてのみ使用すること。
8.2 本剤投与中は定期的に白血球数をモニタリングし、白血球数が50,000/mm3を超えた場合には本剤投与の可否を慎重に判断するとともに、適切な処置を行うこと。
8.3 血小板減少症があらわれることがあるので、本剤投与中は定期的に血小板数をモニタリングし、異常が認められた場合には適切な処置を行うこと。
8.4 ショック、アナフィラキシーを含むアレルギー反応及び過敏症があらわれることがあり、特に本剤の初回投与時に多く認められている。[11.1.1参照]
8.5 脾腫、脾破裂があらわれることがあるので、血液学的検査値の推移に留意するとともに、腹部超音波検査等により観察を十分に行うこと。
9. 特定の背景を有する患者に関する注意
9.2 腎機能障害患者
9.2.1 中等度以上の腎機能障害のある患者
中等度以上の腎機能障害(クレアチニンクリアランス(CLcr)50mL/分以下)のある患者では、本剤の血中濃度が上昇するとの報告があるため、減量を考慮するとともに、患者の状態をより慎重に観察し、有害事象の発現に十分注意すること。[16.6.1参照]
9.4 生殖能を有する者
妊娠する可能性のある女性には、本剤投与中及び最終投与後1週間において避妊する必要性及び適切な避妊法について説明すること。[9.5参照]
9.5 妊婦
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。本剤の乳汁中への移行は検討されていない。
9.7 小児等
小児等を対象とした臨床試験は実施していない。
9.8 高齢者
患者の状態を十分に観察しながら慎重に投与すること。一般に高齢者では生理機能が低下している。
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 ショック、アナフィラキシー(いずれも頻度不明)[8.4参照]
11.1.2 脾腫(頻度不明)、脾破裂(頻度不明)
脾臓の急激な腫大が認められた場合には投与を中止するなど適切な処置を行うこと。
「重大な副作用」及び「その他の副作用」の発現頻度は多発性骨髄腫及び非ホジキンリンパ腫を対象とした海外第3相臨床試験における副作用(全Grade)の集計に基づく。なお、これら以外の試験あるいは海外市販後に認められた副作用は「頻度不明」とした。
11.2 その他の副作用
| 5%以上 | 1〜5%未満 | 1%未満 | 頻度不明 | |
| 精神神経系 | 錯感覚、頭痛 | 不眠症、浮動性めまい | 悪夢 | 異常な夢、血管迷走神経性反応(起立性低血圧、失神) |
| 消化器 | 下痢、悪心 | 鼓腸、腹痛、嘔吐、腹部膨満、腹部不快感、便秘、消化不良、口内乾燥、口の感覚鈍麻 | ||
| 皮膚 | 多汗症、紅斑 | |||
| 血液 | 白血球増加症 | |||
| その他 | 注射部位反応、疲労 | 関節痛、筋骨格痛、倦怠感 |
14. 適用上の注意
14.1 薬剤調製前の注意
バイアル内に微粒子や変色がないか目視で確認し、異常が認められた場合はそのバイアルは使用しないこと。
14.2 薬剤調製時の注意
本剤のバイアルは1回使い切りである。バイアル中の未使用残液は適切に廃棄すること。(本剤は保存剤を含有していない。)
14.3 薬剤投与時の注意
皮下注射にのみ使用すること。
15. その他の注意
15.1 臨床使用に基づく情報
海外の製造販売後において、本剤とG-CSF製剤を投与した急性骨髄性白血病患者及び多発性骨髄腫患者で、循環血中の腫瘍細胞の増加が認められたとの報告がある。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
日本人健康成人18例(各用量6例)にプレリキサホル0.16、0.24及び0.4mg/kg注1)を単回皮下投与したときのプレリキサホルの血漿中濃度推移及び薬物動態パラメータは以下のとおりであった。曝露量(Cmax及びAUC)は、0.16〜0.4mg/kgでほぼ用量に比例して増加した1)。
注1)本剤の承認された通常1回用量は0.24mg/kgである。[6.参照]
プレリキサホルを単回皮下投与したときの血漿中濃度推移
| 投与量(mg/kg) | Cmax(ng/mL) | Tmax注2)(hr) | AUC0-24hr(ng・hr/mL) | t1/2z(hr) |
| 0.16 | 401±46.9 | 0.50(0.50-0.50) | 1740±276 | 5.56±1.30 |
| 0.24 | 685±132 | 0.50(0.50-0.50) | 2690±319 | 5.94±0.777 |
| 0.4 | 1020±92.1 | 0.50(0.25-1.00) | 4600±413 | 5.49±0.522 |
16.1.2 反復投与
16.3 分布
16.3.1 分布容積
日本人健康成人18例(各用量6例)においてプレリキサホル0.16、0.24及び0.4mg/kg注4)を単回皮下投与したときの平均分布容積(Vz/F)は、38.0〜40.3Lであった1)。
16.3.2 蛋白結合率
in vitro試験の結果、プレリキサホル(1〜10μg/mL)のヒト血漿タンパク結合率は37.0〜58.0%であった3)。
注4)本剤の承認された通常1回用量は0.24mg/kgである。[6.参照]
16.4 代謝
ヒト肝ミクロソーム又はヒト肝細胞を用いた試験において、プレリキサホルの代謝は認められなかった4)。
16.5 排泄
本剤は主に尿中に排泄される。
腎機能が正常な健康成人にプレリキサホル0.24mg/kgを単回皮下投与したとき、投与24時間後までに投与量の約70%が未変化体として尿中に排泄された(外国人データ)5)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
外国人腎機能障害患者(軽度[CLcr:51〜80mL/分]、中等度[CLcr:31〜50mL/分]、重度[CLcr:<31mL/分])にプレリキサホル0.24mg/kgを単回皮下投与したときのプレリキサホルの薬物動態パラメータは以下のとおりであった。プレリキサホルのCLは腎機能障害の程度に伴い低下し、CLとCLcrとの間に正の相関関係が認められた。
軽度、中等度及び重度腎機能障害患者における、投与量で補正していないAUC0-24hrの健康被験者に対する最小二乗平均の比はそれぞれ106.6%、132.3%及び138.8%であった5)。[9.2.1参照]
軽度、中等度及び重度腎機能障害患者における、投与量で補正していないAUC0-24hrの健康被験者に対する最小二乗平均の比はそれぞれ106.6%、132.3%及び138.8%であった5)。[9.2.1参照]
| \ | Cmax(ng/mL) | Tmax注5)(hr) | AUC0-24hr(ng・hr/mL) | t1/2(hr) | CL/F(mL/hr) |
| 対照(健康被験者6例) | 980±196 | 0.559(0.50-1.02) | 5070±979 | 4.87±0.562 | 4380±821 |
| 軽度(5例) | 739±76.1 | 0.500(0.50-1.00) | 5410±1070 | 7.80±2.15 | 3500±1690 |
| 中等度(6例) | 936±280 | 0.500(0.25-1.00) | 6780±1660 | 12.1±2.06 | 2420±1110 |
| 重度(6例) | 861±193 | 0.750(0.50-1.00) | 6990±1010 | 15.8±5.79 | 1820±380 |
17. 臨床成績
17.1 有効性及び安全性に関する試験
<多発性骨髄腫に対する自家末梢血幹細胞移植>
17.1.1 国内第2相臨床試験
自家末梢血幹細胞移植に適格な多発性骨髄腫患者を対象に、G-CSF製剤(フィルグラスチム400μg/m2)+本剤(0.24mg/kg)の有効性及び安全性をG-CSF製剤単独と比較した第2相試験を実施した。なお、本剤の投与は末梢血幹細胞採取実施の9〜12時間前に行うこととされた。有効性解析対象集団14例(G-CSF製剤+本剤群7例、G-CSF製剤単独群7例)において、主要評価項目であるCD34陽性細胞数(末梢血幹細胞採取2日以内で6×106cells/kg以上)に到達した患者の割合、並びに副次評価項目であるCD34陽性細胞数(末梢血幹細胞採取4日以内で2×106cells/kg以上)に到達した患者の割合及び6×106cells/kg到達までの日数は、下表のとおりであった6)。
| 有効性評価項目 | G-CSF製剤+本剤群 (7例) | G-CSF製剤単独群 (7例) |
| 2日以内6×106cells/kg以上 | 5(71.4%) | 0(0%) |
| 4日以内2×106cells/kg以上 | 7(100%) | 6(85.7%) |
| 6×106cells/kg到達までの日数 | 2.0注1) | NC注2) |
本剤とG-CSF製剤を併用投与した7例中6例(85.7%)に副作用が認められた。副作用は、背部痛5例(71.4%)、頭痛及び下痢各2例(28.6%)、動悸、腹部不快感、腹痛、関節痛、筋骨格痛及び四肢痛各1例(14.3%)であった。
17.1.2 海外第3相臨床試験
自家末梢血幹細胞移植に適格な多発性骨髄腫患者を対象に、G-CSF製剤(フィルグラスチム10μg/kg)+本剤(0.24mg/kg)の有効性及び安全性をG-CSF製剤+プラセボと比較した第3相試験を実施した。なお、本剤の投与は末梢血幹細胞採取実施の10〜11時間前に行うこととされた。ITT集団302例(G-CSF製剤+本剤群148例、G-CSF製剤+プラセボ群154例)において、主要評価項目であるCD34陽性細胞数(末梢血幹細胞採取2日以内で6×106cells/kg以上)に到達した患者の割合、並びに副次評価項目であるCD34陽性細胞数(末梢血幹細胞採取4日以内で2×106cells/kg以上)に到達した患者の割合及び6×106cells/kg到達までの日数は、下表のとおりであった7)。
| 有効性評価項目 | G-CSF製剤+本剤群 (148例) | G-CSF製剤+プラセボ群 (154例) | P値 |
| 2日以内6×106cells/kg以上 | 106(71.6%) | 53(34.4%) | <0.001注3) |
| 4日以内2×106cells/kg以上 | 141(95.3%) | 136(88.3%) | 0.031注3) |
| 6×106cells/kg到達までの日数 | 1.0注4) | 4.0注4) | − |
| 2.539注5) | / | <0.001注6) |
本剤とG-CSF製剤を併用投与した147例中95例(64.6%)に副作用が認められた。主な副作用は、注射部位紅斑30例(20.4%)、下痢27例(18.4%)、悪心24例(16.3%)、骨痛14例(9.5%)、疲労12例(8.2%)、錯感覚11例(7.5%)等であった。
<非ホジキンリンパ腫に対する自家末梢血幹細胞移植>
17.1.3 国内第2相臨床試験
自家末梢血幹細胞移植に適格な非ホジキンリンパ腫患者を対象に、G-CSF製剤(フィルグラスチム400μg/m2)+本剤(0.24mg/kg)の有効性及び安全性をG-CSF製剤単独と比較した第2相試験を実施した。なお、本剤の投与は末梢血幹細胞採取実施の9〜12時間前に行うこととされた。有効性解析対象集団32例(G-CSF製剤+本剤群16例、G-CSF製剤単独群16例)において、主要評価項目であるCD34陽性細胞数(末梢血幹細胞採取4日以内で5×106cells/kg以上)に到達した患者の割合、並びに副次評価項目であるCD34陽性細胞数(末梢血幹細胞採取4日以内で2×106cells/kg以上)に到達した患者の割合及び5×106cells/kg到達までの日数は、下表のとおりであった8)。
| 有効性評価項目 | G-CSF製剤+本剤群 (16例) | G-CSF製剤単独群 (16例) |
| 4日以内5×106cells/kg以上 | 9(56.3%) | 1(6.3%) |
| 4日以内2×106cells/kg以上 | 15(93.8%) | 5(31.3%) |
| 5×106cells/kg到達までの日数 | 3.5注7) | NC注8) |
本剤とG-CSF製剤を併用投与した16例中12例(75.0%)に副作用が認められた。副作用は、背部痛9例(56.3%)、下痢及び悪心各3例(18.8%)、頭痛及び関節痛各2例(12.5%)、高尿酸血症、潮紅、ほてり、口の感覚鈍麻、門脈ガス血症、注射部位そう痒感、疲労、発熱、血中乳酸脱水素酵素増加及び血小板数減少各1例(6.3%)であった。
17.1.4 海外第3相臨床試験
自家末梢血幹細胞移植に適格な非ホジキンリンパ腫患者を対象に、G-CSF製剤(フィルグラスチム10μg/kg)+本剤(0.24mg/kg)の有効性及び安全性をG-CSF製剤+プラセボと比較した第3相試験を実施した。なお、本剤の投与は末梢血幹細胞採取実施の10〜11時間前に行うこととされた。ITT集団298例(G-CSF製剤+本剤群150例、G-CSF製剤+プラセボ群148例)において、主要評価項目であるCD34陽性細胞数(末梢血幹細胞採取4日以内で5×106cells/kg以上)に到達した患者の割合、並びに副次評価項目であるCD34陽性細胞数(末梢血幹細胞採取4日以内で2×106cells/kg以上)に到達した患者の割合及び5×106cells/kg到達までの日数は、下表のとおりであった9)。
| 有効性評価項目 | G-CSF製剤+本剤群 (150例) | G-CSF製剤+プラセボ群 (148例) | P値 |
| 4日以内5×106cells/kg以上 | 89(59.3%) | 29(19.6%) | <0.001注9) |
| 4日以内2×106cells/kg以上 | 130(86.7%) | 70(47.3%) | <0.001注10) |
| 5×106cells/kg到達までの日数 | 3.0注11) | NC注12) | − |
| 3.643注13) | / | <0.001注14) |
本剤とG-CSF製剤を併用投与した150例中98例(65.3%)に副作用が認められた。主な副作用は、下痢56例(37.3%)、注射部位紅斑44例(29.3%)、悪心26例(17.3%)、頭痛16例(10.7%)、骨痛14例(9.3%)、注射部位そう痒感12例(8.0%)、錯感覚10例(6.7%)等であった。
18. 薬効薬理
18.1 作用機序
骨髄の間質細胞表面に発現するSDF-1は、CXCR4を発現している造血幹細胞の骨髄への生着に関与していると考えられている。プレリキサホルはCXCR4に結合し、CXCR4とSDF-1との結合を阻害することにより、骨髄から末梢血中への造血幹細胞の動員を促進すると考えられる10)。
18.2 薬理作用
19. 有効成分に関する理化学的知見
19.1. プレリキサホル
| 一般的名称 | プレリキサホル |
|---|---|
| 一般的名称(欧名) | Plerixafor |
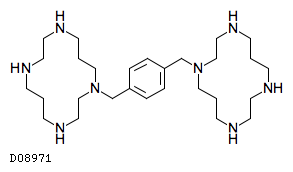
| 化学名 | 1,1'-(1,4-Phenylenebismethylene)bis(1,4,8,11-tetraazacyclotetradecane) |
| 分子式 | C28H54N8 |
| 分子量 | 502.78 |
| 融点 | 131.5℃ |
| 物理化学的性状 | 本品は白色〜灰白色の固体である。 本品はエタノール(99.5)又はメタノールにやや溶けやすく、水に溶けにくい。 |
| 分配係数 | Log P=0.94(1-オクタノール/水) |
| KEGG DRUG |
|
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
24mg/1.2mL×1バイアル
23. 主要文献
- 社内資料:日本人健康成人における薬物動態(2016年12月19日承認、CTD2.7.2.2)
- 社内資料:外国人患者における蓄積性(2016年12月19日承認、CTD2.7.2.3)
- 社内資料:血漿タンパク結合に関する検討(2016年12月19日承認、CTD2.6.4.4)
- 社内資料:代謝に関する検討(in vitro)(2016年12月19日承認、CTD2.7.2.2)
- 社内資料:外国人腎機能障害患者における薬物動態(2016年12月19日承認、CTD2.7.2.2)
- 社内資料:国内第II相臨床試験(多発性骨髄腫)(2016年12月19日承認、CTD2.7.6.2)
- DiPersio J F,et al., Blood., 113 (23), 5720-26, (2009) »PubMed
- 社内資料:国内第II相臨床試験(非ホジキンリンパ腫)(2016年12月19日承認、CTD2.7.6.2)
- DiPersio J F,et al., J Clin Oncol., 27 (28), 4767-73, (2009) »PubMed
- Fricker S P, Transfus Med Hemother., 40 (4), 237-45, (2013) »PubMed
- Fricker S P,et al., Biochem Pharmacol., 72 (5), 588-96, (2006) »PubMed
- Broxmeyer H E,et al., J Exp Med., 201 (8), 1307-18, (2005) »PubMed
- Burroughs L,et al., Blood., 106 (12), 4002-8, (2005) »PubMed
24. 文献請求先及び問い合わせ先
文献請求先
サノフィ株式会社
くすり相談室
〒163-1488
東京都新宿区西新宿三丁目20番2号
電話:フリーダイヤル 0120-109-905
製品情報問い合わせ先
サノフィ株式会社
くすり相談室
〒163-1488
東京都新宿区西新宿三丁目20番2号
電話:フリーダイヤル 0120-109-905
26. 製造販売業者等
26.1 製造販売元
サノフィ株式会社
〒163-1488
東京都新宿区西新宿三丁目20番2号