医薬品情報
| 総称名 | オテズラ |
|---|---|
| 一般名 | アプレミラスト |
| 欧文一般名 | Apremilast |
| 製剤名 | アプレミラスト錠 |
| 薬効分類名 | PDE4阻害剤 |
| 薬効分類番号 | 3999 |
| ATCコード | L04AA32 |
| KEGG DRUG |
D08860
アプレミラスト
|
| JAPIC | 添付文書(PDF) |
添付文書情報2025年3月 改訂(第4版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| オテズラ錠10mg | Otezla Tablets | アムジェン | 3999042F1025 | 284.4円/錠 | 劇薬, 処方箋医薬品 |
| オテズラ錠20mg | Otezla Tablets | アムジェン | 3999042F2021 | 568.7円/錠 | 劇薬, 処方箋医薬品 |
| オテズラ錠30mg | Otezla Tablets | アムジェン | 3999042F3028 | 853.1円/錠 | 劇薬, 処方箋医薬品 |
2. 禁忌
次の患者には投与しないこと
2.1 本剤の成分に対し過敏症の既往歴のある患者
2.2 妊婦又は妊娠している可能性のある女性[9.5参照]
4. 効能または効果
5. 効能または効果に関連する注意
<局所療法で効果不十分な尋常性乾癬、乾癬性関節炎>
5.1 以下のいずれかを満たす尋常性乾癬又は乾癬性関節炎患者に投与すること。
・ステロイド外用剤等で十分な効果が得られず、皮疹が体表面積の10%以上に及ぶ患者
・難治性の皮疹又は関節症状を有する患者
<局所療法で効果不十分な掌蹠膿疱症>
5.2 中等症から重症の膿疱・小水疱病変を有する患者に投与すること。
6. 用法及び用量
通常、成人にはアプレミラストとして以下のとおり経口投与し、6日目以降はアプレミラストとして1回30mgを1日2回、朝夕に経口投与する。
| 1日目 | 2日目 | 3日目 | 4日目 | 5日目 | 6日目以降 | |||||
| 朝 | 朝 | 夕 | 朝 | 夕 | 朝 | 夕 | 朝 | 夕 | 朝 | 夕 |
| 10mg | 10mg | 10mg | 10mg | 20mg | 20mg | 20mg | 20mg | 30mg | 30mg | 30mg |
7. 用法及び用量に関連する注意
<効能共通>
7.1 投与開始時に漸増投与を行わなかった場合、悪心、下痢、嘔吐等の発現率が高いことが示されているため、「用法・用量」を遵守すること。[11.2参照]
<局所療法で効果不十分な尋常性乾癬、乾癬性関節炎>
7.3 本剤による治療反応は、通常投与開始から24週以内に得られる。24週以内に治療反応が得られない場合は、本剤の治療計画の継続を慎重に再考すること。
<局所療法で効果不十分な掌蹠膿疱症>
7.4 本剤による治療反応は、通常投与開始から16週以内に得られる。16週以内に治療反応が得られない場合は、本剤の治療計画の継続を慎重に再考すること。
8. 重要な基本的注意
本剤の投与は適応疾患の治療に十分な知識・経験を持つ医師のもとで行うこと。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 感染症の患者、感染症が疑われる又は再発性感染症の既往歴のある患者
感染症を悪化又は顕在化させるおそれがある。
9.2 腎機能障害患者
9.2.1 重度の腎機能障害のある患者(Cockcroft-Gault式によるクレアチニンクリアランス値が30mL/min未満)
9.4 生殖能を有する者
妊娠する可能性のある女性に対しては、本剤投与前に問診等により妊娠していないことを確認し、本剤が胚胎児毒性のリスクを有する可能性があることを説明した上で投与を開始すること。また、本剤投与中及び最終投与後48時間において避妊する必要性及び適切な避妊法について説明すること。[9.5参照]
9.5 妊婦
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。本剤のヒトにおける乳汁への移行は不明であるが、本剤を投与した動物試験(マウス)で乳汁への移行が報告されている。
9.7 小児等
小児等は臨床試験では除外されている。
9.8 高齢者
感染症、下痢、悪心、嘔吐等の副作用の発現に留意し、患者の状態を十分に観察しながら、慎重に投与すること。一般に生理機能が低下している。
10. 相互作用
相互作用序文
本剤は主にCYP3A4で代謝される。[16.4参照]
薬物代謝酵素用語
CYP3A4
10.2 併用注意
| CYP3A4酵素誘導作用を有する薬剤 (リファンピシン、フェノバルビタール、カルバマゼピン、フェニトイン等) [16.7.1参照] | 本剤の効果の減弱に注意すること。 | 本剤の血漿中濃度が減少すると考えられる。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 重篤な感染症(0.7%)
ウイルス、細菌、真菌等による重篤な感染症があらわれることがある。
11.1.2 重篤な過敏症(0.1%未満)
アナフィラキシー等の過敏症があらわれることがある。
11.1.3 重度の下痢(頻度不明)
11.2 その他の副作用
| 5%以上 | 1〜5%未満 | 1%未満 | |
| 感染症・寄生虫症 | 上気道感染、上咽頭炎、気管支炎、副鼻腔炎 | 尿路感染、咽頭炎、ウイルス性上気道感染 | |
| 胃腸障害 | 下痢、悪心 | 嘔吐、腹痛、消化不良、軟便、上腹部痛、腹部不快感、排便回数増加、胃食道逆流性疾患 | 腹部膨満 |
| 神経系障害・精神障害 | 頭痛 | 緊張性頭痛、片頭痛 | 浮動性めまい、不眠症、うつ病 |
| 代謝・栄養障害 | 食欲減退、体重減少 | ||
| その他 | 疲労、乾癬 | 咳嗽、高血圧、そう痒症、発疹、背部痛、過敏症 |
14. 適用上の注意
14.1 薬剤交付時の注意
14.1.1 PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔を起こして縦隔洞炎等の重篤な合併症を併発することがある。
14.1.2 錠剤を噛み砕いたり、割ったりせずに服用するよう指導すること。
15. その他の注意
15.1 臨床使用に基づく情報
<局所療法で効果不十分な尋常性乾癬、乾癬性関節炎>
国内臨床試験(254例)において、うつ病及び自殺関連事象は報告されなかった。
海外臨床試験(併合)のプラセボ対照期において、うつ病は、プラセボ群1411例中8例(0.6%)、本剤30mg1日2回投与群1668例中17例(1.0%)に認められ、このうちプラセボ群2例(0.1%)、本剤30mg1日2回投与群4例(0.2%)については本剤との因果関係は否定されなかった。また自殺関連事象は、プラセボ群1411例中1例(0.1%:自殺既遂)、本剤30mg1日2回投与群1668例中2例(0.1%:自殺企図、自殺念慮各1例)に認められ、いずれも本剤との因果関係は否定されている。
海外臨床試験(併合)の本剤の全投与期間において、うつ病は、本剤30mg1日2回投与群2357例中63例(2.7%)に認められ、このうち10例(0.4%)については本剤との因果関係は否定されなかった。また自殺関連事象は、本剤30mg1日2回投与群2357例中3例(0.1%:自殺企図2例、自殺念慮1例)に認められ、いずれも本剤との因果関係は否定されている。
<局所療法で効果不十分な掌蹠膿疱症>
国内第III相試験(174例)において、うつ病及び自殺関連事象は報告されなかった。
<局所療法で効果不十分なベーチェット病による口腔潰瘍>
国際共同第III相試験のプラセボ対照期において、うつ病は、プラセボ群103例中1例(1.0%)、本剤30mg1日2回投与群104例中1例(1.0%)に認められ、このうちプラセボ群1例については治験薬との因果関係は否定されなかった。本剤全投与期間においてうつ病は本剤30mg1日2回投与群187例中2例(1.1%)に認められ、いずれも本剤との因果関係は否定されている。また、自殺関連事象は、プラセボ対照期及び本剤全投与期間のいずれでも認められなかった。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
健康成人に本剤20mg注)及び40mg注)を単回経口投与したときの本剤の血漿中濃度推移と薬物動態パラメータは、以下のとおりであった。
単回経口投与したときの血漿中濃度推移(平均値±標準偏差)
| 20mg注) (12例) | 40mg注) (12例) | |
| AUCt(ng・h/mL) | 1515(21.9) | 2921(17.2) |
| AUC∞(ng・h/mL) | 1532(21.2) | 2943(17.1) |
| Cmax(ng/mL) | 211(31.3) | 343(25.9) |
| tmaxa(h) | 2.50(1.00,6.00) | 3.50(2.00,6.00) |
| t1/2(h) | 5.44(15.8) | 5.32(16.3) |
| CL/F(L/h) | 13.1(21.2) | 13.6(17.1) |
| Vz/F(L) | 102(27.2) | 104(28.4) |
注)本剤の漸増投与後の承認用法・用量は「1回30mgを1日2回、朝夕に経口投与」である。
16.1.2 反復投与
尋常性乾癬患者及びベーチェット病患者に本剤30mgを1日2回反復経口投与したときの本剤の定常状態における薬物動態パラメータは、以下のとおりであった。
| 尋常性乾癬患者 (20例、投与20週時) | ベーチェット病患者 (7例、投与16週時) | |
| AUCτ(ng・h/mL) | 2397(39.5) | 2071(49.5) |
| Cmax(ng/mL) | 374(32.0) | 374.2(31.3) |
| tmaxa(h) | 2.00(0.98,4.00) | 1.08(1.00,2.00) |
| t1/2(h) | 4.06(23.6)b | 4.23(26.9) |
| CL/F(L/h) | 12.9(34.1)b | 14.45(49.5) |
| Vz/F(L) | 83.1(32.2)b | 88.3(46.1) |
尋常性乾癬、ベーチェット病及び掌蹠膿疱症の日本人成人患者に本剤30mgを1日2回反復経口投与したときのトラフ濃度の幾何平均値(%変動係数)は、それぞれ116ng/mL(74.0)[20週後、20例]、88ng/mL(76.0)[16週後、7例]及び160ng/mL(76.0)[16週後、49例]であった。
健康成人(6例)に本剤40mg注)を1日2回反復経口投与したとき、本剤は速やかに吸収され、約2.5時間(tmax:中央値)でCmaxに達した。健康成人に本剤50mg注)を1日2回反復経口投与(6例)又は80mg注)を1日1回反復経口投与(9例)したとき、AUC∞及びCmaxは用量依存的に増加した(外国人データ)。
また、中等症〜重症の尋常性乾癬患者に本剤10mg注)(7例)、20mg注)(5例)及び30mg(3例)を1日2回反復経口投与したとき、本剤は速やかに吸収され、約2時間(tmax:中央値)でCmaxに達した。その後、血漿中濃度は減少し、消失半減期は4.93〜6.56時間であった。なお、AUCτ及びCmaxは用量依存的に増加した(外国人データ)。
注)本剤の漸増投与後の承認用法・用量は「1回30mgを1日2回、朝夕に経口投与」である。
16.2 吸収
16.2.1 バイオアベイラビリティ
健康成人(12例)に本剤20mg注)を経口投与したときの吸収の絶対バイオアベイラビリティは約73%であった(外国人データ)。
注)本剤の漸増投与後の承認用法・用量は「1回30mgを1日2回、朝夕に経口投与」である。
16.2.2 食事の影響
健康成人(46例)に本剤30mgを食後に単回経口投与したとき、AUC(AUC∞及びAUCt)及びCmaxへの食事の影響は認められなかった(外国人データ)。
16.3 分布
16.3.1 血漿蛋白結合率
アプレミラストのヒト血漿における蛋白結合率は約68%であった。
16.4 代謝
健康成人において、放射性標識したアプレミラストを経口投与したとき、血漿中総放射能に対して未変化体が約45%、次いでO-脱メチル化アプレミラストのグルコロニド抱合体である不活性代謝物が約39%認められた(外国人データ)。
アプレミラストはチトクロムP450酸化代謝に続くグルクロン酸抱合及びチトクロムP450以外の加水分解により代謝されると考えられ、in vitro試験において、アプレミラストの代謝に関与するチトクロムP450は主にCYP3A4であることが示唆されたが、CYP1A2及びCYP2A6の関与も認められた。[10.参照]
アプレミラストはチトクロムP450酸化代謝に続くグルクロン酸抱合及びチトクロムP450以外の加水分解により代謝されると考えられ、in vitro試験において、アプレミラストの代謝に関与するチトクロムP450は主にCYP3A4であることが示唆されたが、CYP1A2及びCYP2A6の関与も認められた。[10.参照]
16.5 排泄
健康成人において、放射性標識したアプレミラストを経口投与したとき、尿中及び糞便中における投与量に対する放射能回収率は、それぞれ約58%及び39%で、未変化体アプレミラストの回収率は、尿中及び糞便中で、それぞれ約3%及び4%であった(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
腎機能障害を有する被験者に本剤30mgを単回投与したときの薬物動態パラメータは以下のとおりであり、重度の腎機能障害を有する被験者では、正常な腎機能を有する被験者と比較してAUC∞及びCmaxは、それぞれ約88%及び42%増加した(外国人データ)。[7.2、9.2.1参照]
| 腎機能eGFR | 軽度 (8例) | 軽度対照 (8例) | 中等度 (8例) | 中等度対照 (8例) | 重度 (8例) | 重度対照 (7例) |
| AUC∞(ng・h/mL) | 2975(21) | 3464(19) | 3466(67) | 2838(24) | 5425(53) | 2879(18) |
| Cmax(ng/mL) | 265(30) | 250(17) | 182(47) | 208(32) | 366(35) | 255(40) |
| tmaxa(h) | 3.0(2.0,4.0) | 3.0(2.0,4.1) | 3.5(0.5,8.0) | 2.0(1.0,6.0) | 3.0(1.0,6.0) | 3.0(2.0,4.0) |
| t1/2(h) | 8.4(19) | 8.1(24) | 10.5(40) | 8.3(24) | 11.8(18) | 9.4(18) |
16.6.2 肝機能障害患者
アプレミラストとその主要代謝物、O-脱メチル化アプレミラストのグルコロニド抱合体の薬物動態について、中等度(Child-Pugh7〜9)又は重度(Child-Pugh10〜13)の肝機能障害を有する被験者で影響は認められなかった(外国人データ)。
16.6.3 高齢者
本剤を高齢の健康被験者(65〜85歳)に投与したとき、AUC(AUC∞及びAUCt)及びCmaxは非高齢の健康被験者(18〜55歳)と比べてそれぞれ約13%及び6%増加した(外国人データ)。
16.6.4 女性
本剤を女性の健康被験者に投与したとき、AUC∞及びCmaxは男性の健康被験者と比べてそれぞれ約31%及び8%増加した(外国人データ)。
16.7 薬物相互作用
16.7.1 リファンピシン
本剤とリファンピシンを併用したとき、アプレミラストのAUC(AUC∞及びAUCt)及びCmaxはそれぞれ約72%及び43%減少した(外国人データ)。[10.2参照]
16.7.2 ケトコナゾール
本剤とケトコナゾールを併用したとき、アプレミラストのAUC∞及びCmaxはそれぞれ約36%及び5%増加した(外国人データ)。
16.7.3 メトトレキサート
本剤とメトトレキサートを併用したとき、アプレミラストのAUCτ及びCmaxはそれぞれ約0.7%及び5%減少した(外国人データ)。
17. 臨床成績
17.1 有効性及び安全性に関する試験
<局所療法で効果不十分な尋常性乾癬、乾癬性関節炎>
17.1.1 国内後期第II相試験
体表面積(BSA)10%以上及びPsoriasis Area and Severity Index(PASI)スコア12以上の中等症〜重症の尋常性乾癬及び乾癬性関節炎患者を対象としたランダム化プラセボ対照二重盲検並行群間比較試験(PSOR-011試験)の結果は以下のとおりであった。
主要評価項目である投与16週時のPASI-75及び副次評価項目である医師による静的全般評価(Static Physician Global Assessment:sPGA)が0(消失)又は1(ほぼ消失)を達成した患者の割合において、本剤30mg1日2回投与群はプラセボ群と比べて有意に高かった1)。
主要評価項目である投与16週時のPASI-75及び副次評価項目である医師による静的全般評価(Static Physician Global Assessment:sPGA)が0(消失)又は1(ほぼ消失)を達成した患者の割合において、本剤30mg1日2回投与群はプラセボ群と比べて有意に高かった1)。
| 20mg注)群 | 30mg群 | プラセボ群 | プラセボ群との群間差 [95%CI] p値a,b | ||
| 20mg注)群 | 30mg群 | ||||
| PASI-75達成率 | 23.5 (20/85) | 28.2 (24/85) | 7.1 (6/84) | 16.4 [5.8,27.0] p=0.0032 | 21.1 [10.1,32.1] p=0.0003 |
| sPGA(0又は1)達成率c | 23.9 (17/71) | 29.6 (21/71) | 8.8 (6/68) | 15.1 [3.1,27.1] | 20.8 [8.2,33.3] |
全投与期間中(0〜68週)に本剤30mgを1日2回投与された安全性評価症例120例中37例(30.8%)で副作用が認められた。主な副作用は、下痢11例(9.2%)、腹部不快感7例(5.8%)、鼻咽頭炎5例(4.2%)、乾癬3例(2.5%)、腹部膨満3例(2.5%)であった。
注)本剤の漸増投与後の承認用法・用量は「1回30mgを1日2回、朝夕に経口投与」である。
17.1.2 海外第III相試験
BSA10%以上、PASIスコア12以上、sPGAスコア3以上の中等症〜重症の尋常性乾癬及び乾癬性関節炎患者を対象としたランダム化プラセボ対照二重盲検並行群間比較試験(PSOR-008試験)の結果は以下のとおりであった。
主要評価項目である投与16週時のPASI-75及び副次評価項目であるsPGAが0(消失)又は1(ほぼ消失)を達成した患者の割合において、本剤30mg1日2回投与群はプラセボ群と比べて有意に高かった2)3)。
主要評価項目である投与16週時のPASI-75及び副次評価項目であるsPGAが0(消失)又は1(ほぼ消失)を達成した患者の割合において、本剤30mg1日2回投与群はプラセボ群と比べて有意に高かった2)3)。
| 30mg群 | プラセボ群 | 群間差 [95%CI] p値a | |
| PASI-75達成率 | 33.1 (186/562) | 5.3 (15/282) | 27.8 [23.1,32.5] p<0.0001 |
| sPGA(0又は1)達成率 | 21.7 (122/562) | 3.9 (11/282) | 17.8 [13.7,21.9] |
本剤の全投与期間中(0〜52週)に安全性評価症例804例中340例(42.3%)で副作用が認められた。主な副作用は、下痢128例(15.9%)、悪心104例(12.9%)、緊張性頭痛39例(4.9%)、頭痛31例(3.9%)、上気道感染31例(3.9%)、排便回数増加22例(2.7%)、鼻咽頭炎21例(2.6%)、嘔吐21例(2.6%)、腹部不快感19例(2.4%)、消化不良17例(2.1%)、食欲減退16例(2.0%)であった。
17.1.3 海外第III相試験
BSA10%以上、PASIスコア12以上、sPGAスコア3以上の中等症〜重症の尋常性乾癬及び乾癬性関節炎患者を対象としたランダム化プラセボ対照二重盲検並行群間比較試験(PSOR-009試験)の結果は以下のとおりであった。
主要評価項目である投与16週時のPASI-75及び副次評価項目であるsPGAが0(消失)又は1(ほぼ消失)を達成した患者の割合において、本剤30mg1日2回投与群はプラセボ群と比べて有意に高かった4)5)。
主要評価項目である投与16週時のPASI-75及び副次評価項目であるsPGAが0(消失)又は1(ほぼ消失)を達成した患者の割合において、本剤30mg1日2回投与群はプラセボ群と比べて有意に高かった4)5)。
| 30mg群 | プラセボ群 | 群間差 [95%CI] p値a | |
| PASI-75達成率 | 28.8 (79/274) | 5.8 (8/137) | 23.0 [16.3,29.6] p<0.0001 |
| sPGA(0又は1)達成率 | 20.4 (56/274) | 4.4 (6/137) | 16.1 [10.2,21.9] |
本剤の全投与期間中(0〜52週)に安全性評価症例380例中151例(39.7%)で副作用が認められた。主な副作用は、悪心54例(14.2%)、下痢45例(11.8%)、緊張性頭痛17例(4.5%)、頭痛13例(3.4%)、嘔吐12例(3.2%)、腹痛10例(2.6%)、消化不良9例(2.4%)、鼻咽頭炎8例(2.1%)であった。
17.1.4 海外第III相試験
疾患修飾性抗リウマチ薬(DMARD)による前治療、又はそれらによる治療にもかかわらず活動性を示す乾癬性関節炎患者(3個以上の腫脹関節及び3個以上の圧痛関節)を対象としたランダム化プラセボ対照二重盲検並行群間比較試験(PSA-002試験)の結果は以下のとおりであった。本試験では本剤とメトトレキサートを含む低分子DMARDとの併用が可能とされた。
主要評価項目である投与16週時のアメリカリウマチ学会コアセット20%改善(ACR20)を達成した患者の割合において、本剤30mg1日2回投与群はプラセボ群に比べて有意に高かった6)7)8)。
主要評価項目である投与16週時のアメリカリウマチ学会コアセット20%改善(ACR20)を達成した患者の割合において、本剤30mg1日2回投与群はプラセボ群に比べて有意に高かった6)7)8)。
| 30mg群 | プラセボ群 | 群間差a [95%CI] p値b | |
| ACR20改善率 | 38.1 (64/168) | 19.0 (32/168) | 19.0 [9.7,28.3] p=0.0001 |
本剤の全投与期間中(0〜52週)に安全性評価症例245例中108例(44.1%)で副作用が認められた。主な副作用は、下痢40例(16.3%)、悪心29例(11.8%)、頭痛18例(7.3%)、腹痛8例(3.3%)、上腹部痛7例(2.9%)、嘔吐7例(2.9%)、浮動性めまい6例(2.4%)、消化不良6例(2.4%)、胃食道逆流性疾患6例(2.4%)であった。
17.1.5 海外第III相試験
疾患修飾性抗リウマチ薬(DMARD)による前治療、又はそれらによる治療にもかかわらず活動性を示す乾癬性関節炎患者(3個以上の腫脹関節及び3個以上の圧痛関節)を対象としたランダム化プラセボ対照二重盲検並行群間比較試験(PSA-003試験)の結果は以下のとおりであった。本試験では本剤とメトトレキサートを含む低分子DMARDとの併用が可能とされた。
主要評価項目である投与16週時のACR20を達成した患者の割合において、本剤30mg1日2回投与群はプラセボ群に比べて有意に高かった9)。
主要評価項目である投与16週時のACR20を達成した患者の割合において、本剤30mg1日2回投与群はプラセボ群に比べて有意に高かった9)。
| 30mg群 | プラセボ群 | 群間差a [95%CI] p値b | |
| ACR20改善率 | 32.1 (52/162) | 18.9 (30/159) | 13.4 [4.0,22.7] p=0.0060 |
本剤の全投与期間中(0〜52週)に安全性評価症例234例中73例(31.2%)で副作用が認められた。主な副作用は、悪心26例(11.1%)、下痢24例(10.3%)、頭痛15例(6.4%)、消化不良6例(2.6%)、上腹部痛5例(2.1%)、嘔吐5例(2.1%)であった。
17.1.6 海外第III相試験
疾患修飾性抗リウマチ薬(DMARD)による前治療、又はそれらによる治療にもかかわらず活動性を示す乾癬性関節炎患者(3個以上の腫脹関節及び3個以上の圧痛関節)を対象としたランダム化プラセボ対照二重盲検並行群間比較試験(PSA-004試験)の結果は以下のとおりであった。本試験では本剤とメトトレキサートを含む低分子DMARDとの併用が可能とされた。
主要評価項目である投与16週時のACR20を達成した患者の割合において、本剤30mg1日2回投与群はプラセボ群に比べて有意に高かった10)11)。
主要評価項目である投与16週時のACR20を達成した患者の割合において、本剤30mg1日2回投与群はプラセボ群に比べて有意に高かった10)11)。
| 30mg群 | プラセボ群 | 群間差a [95%CI] p値b | |
| ACR20改善率 | 40.7 (68/167) | 18.3 (31/169) | 22.3 [13.0,31.6] p<0.0001 |
本剤の全投与期間中(0〜52週)に安全性評価症例242例中85例(35.1%)で副作用が認められた。主な副作用は、下痢26例(10.7%)、悪心26例(10.7%)、頭痛17例(7.0%)、排便回数増加8例(3.3%)、腹痛5例(2.1%)、食欲減退5例(2.1%)、鼻咽頭炎5例(2.1%)、嘔吐5例(2.1%)であった。
17.1.7 海外第III相試験
低分子DMARDによる前治療歴のない乾癬性関節炎患者を対象としたランダム化プラセボ対照二重盲検並行群間比較試験(PSA-005試験)の結果は以下のとおりであった。
主要評価項目である投与16週時のACR20を達成した患者の割合において、本剤30mg1日2回投与群はプラセボ群に比べて有意に高かった12)。
主要評価項目である投与16週時のACR20を達成した患者の割合において、本剤30mg1日2回投与群はプラセボ群に比べて有意に高かった12)。
| 30mg群 | プラセボ群 | 群間差a [95%CI] p値b | |
| ACR20改善率 | 30.7 (54/176) | 15.9 (28/176) | 14.8 [6.1,23.5] p=0.0010 |
本剤の全投与期間中(0〜24週)に安全性評価症例226例中65例(28.8%)で副作用が認められた。主な副作用は、悪心24例(10.6%)、下痢21例(9.3%)、頭痛9例(4.0%)であった。
<局所療法で効果不十分な掌蹠膿疱症>
17.1.8 国内第III相試験
Palmoplantar Pustulosis Area and Severity Index(PPPASI)総スコアが12以上かつ手掌又は足底にPPPASI重症度スコア2以上の膿疱・小水疱を有する掌蹠膿疱症(PPP)患者を対象としたランダム化プラセボ対照二重盲検並行群間比較試験(20200195試験)の結果は以下のとおりであった。なお、本試験ではPPPに対する治療として投与16週以降は外用療法、光線療法及び全身性抗ヒスタミン薬の併用を可能とした。また、全投与期間中、エトレチナート等の全身療法及び生物製剤の併用を禁止した。
主要評価項目である投与16週時のPPPASI総スコアがベースラインから50%以上改善した患者の割合(PPPASI-50達成率)において、本剤30mg1日2回投与群はプラセボ群と比較して有意に高かった13)。
主要評価項目である投与16週時のPPPASI総スコアがベースラインから50%以上改善した患者の割合(PPPASI-50達成率)において、本剤30mg1日2回投与群はプラセボ群と比較して有意に高かった13)。
| 30mg群 (n=88) | プラセボ群 (n=88) | 群間差 [95%CI]b p値c | |
| PPPASI-50達成率(%) [95%CI] | 67.8 [57.9,77.6] | 35.3 [25.3,45.4] | 32.6 [18.7,46.5] p<0.0001 |
本剤の全投与期間中(0〜52週)に本剤30mgを1日2回投与された安全性評価症例174例中73例(42.0%)で副作用が認められた。主な副作用は、下痢26例(14.9%)、軟便22例(12.6%)、悪心20例(11.5%)、頭痛11例(6.3%)、腹痛6例(3.4%)、腹部不快感5例(2.9%)、食欲減退4例(2.3%)であった。
<局所療法で効果不十分なベーチェット病による口腔潰瘍>
17.1.9 国際共同第III相試験
局所療法で効果不十分なベーチェット病による口腔潰瘍を有する患者を対象としたランダム化プラセボ対照二重盲検並行群間比較試験(BCT-002試験)の結果は以下のとおりであった。
主要評価項目である投与12週時までの口腔潰瘍数のAUCにおいて、本剤30mg1日2回投与群はプラセボ群と比べて統計学的に有意に低かった。また、本剤長期投与時の口腔潰瘍数の経時的推移は下図のとおりであった14)。
主要評価項目である投与12週時までの口腔潰瘍数のAUCにおいて、本剤30mg1日2回投与群はプラセボ群と比べて統計学的に有意に低かった。また、本剤長期投与時の口腔潰瘍数の経時的推移は下図のとおりであった14)。
| 30mg群 | プラセボ群 | ||
| 口腔潰瘍数のAUCW12 | 評価患者数 | 104 | 103 |
| 最小二乗平均 [95%CI] | 129.54 [98.09,160.99] | 222.14 [190.80,253.47] | |
| 群間差 [95%CI]a p値b | −92.60 [−130.59,−54.60] p<0.0001 | ||
国際共同第III相試験における口腔潰瘍数の経時的推移(ITT、実測値)
| 投与後経過(週) | 0/BS | 1 | 2 | 4 | 6 | 8 | 10 | 12 | 16 | 28 | 40 | 52 | 64 | 68/FU | |
| プラセボ/30mg群 | n | 103 | 98 | 97 | 93 | 91 | 86 | 83 | 82 | 83 | 78 | 73 | 70 | 67 | 82 |
| Mean | 3.9 | 2.9 | 2.8 | 2.3 | 2.5 | 2.2 | 1.9 | 2.0 | 0.7 | 0.8 | 0.7 | 1.1 | 0.8 | 2.0 | |
| 30mg群 | n | 104 | 101 | 101 | 101 | 98 | 94 | 94 | 97 | 95 | 92 | 85 | 79 | 75 | 85 |
| Mean | 4.2 | 1.9 | 1.4 | 1.3 | 1.6 | 1.2 | 1.0 | 1.1 | 0.9 | 0.9 | 0.9 | 0.9 | 1.4 | 2.5 | |
BS:ベースライン
FU:追跡調査期
n:評価対象被験者数
Mean:口腔潰瘍数の平均値
プラセボ/30mg群は、12週時に本剤投与に切り替えられた。
いずれの群も投与64週時以降は本剤の投与は行われていない。
BCT-002試験において本剤の全投与期間中(0〜64週)に安全性評価症例187例中93例(49.7%)に副作用が認められた。主な副作用は、下痢58例(31.0%)、悪心23例(12.3%)、頭痛20例(10.7%)、嘔吐9例(4.8%)、上腹部痛7例(3.7%)、腹痛7例(3.7%)であった。
18. 薬効薬理
18.1 作用機序
本剤は、ホスホジエステラーゼ(PDE)4を阻害する低分子の経口PDE4阻害剤で、細胞内で炎症性及び抗炎症メディエーターのネットワークを調節する。PDE4はcAMPに特異的なPDEで、主に炎症性細胞に分布している。本剤は、PDE4を阻害することにより細胞内cAMP濃度を上昇させ、IL-17、TNF-α、IL-23及び他の炎症性サイトカインの発現を制御することにより炎症反応を抑制する。
18.2 In vitroにおける薬理活性
18.2.1 cAMPの加水分解により測定したPDE4活性に対する競合的かつ可逆的な阻害作用を示した(IC50=74nM、Ki=68nM)。また、PDE4A、PDE4B、PDE4C、PDE4Dのいずれのサブタイプに対しても阻害作用を示した15)16)。
18.2.2 ヒト由来精製T細胞において、IL-17等の炎症性サイトカインの産生抑制作用を示した(IL-17産生抑制:IC50=90nM)16)。
18.2.3 ヒト末梢血単核球細胞において、TNF-α等のエンドトキシン誘発性の炎症性サイトカインの産生抑制作用を示した(TNF-α産生抑制:IC50=110nM)。一方、抗炎症サイトカインであるIL-10の産生増加作用を示した15)。
18.3 In vivoにおける薬理活性
19. 有効成分に関する理化学的知見
19.1. アプレミラスト
| 一般的名称 | アプレミラスト |
|---|---|
| 一般的名称(欧名) | Apremilast |
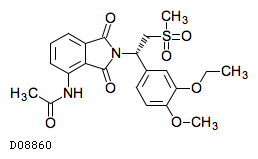
| 化学名 | N-{2-[(1S)-1-(3-Ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide |
| 分子式 | C22H24N2O7S |
| 分子量 | 460.50 |
| 融点 | 約156.1℃ |
| 物理化学的性状 | 白色から淡黄色の粉末である。水にほとんど溶けず、エタノールに溶けにくく、アセトンにやや溶けやすい。アプレミラストはS-エナンチオマーで、20mg/mLのアセトニトリル中で測定するとき、比旋光度は+28.1°である。 |
| KEGG DRUG |
|
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
<オテズラ錠スターターパック>
27錠[(10mg×4錠、20mg×4錠、30mg×19錠)×1パック]
<オテズラ錠30mg>
56錠[14錠(PTP)×4シート]
23. 主要文献
- CC-10004-PSOR-011試験(承認年月日:2016年12月19日、CTD2.7.6.23)
- Papp K,et al., J Am Acad Dermatol., 73, 37-49, (2015) »PubMed »DOI
- CC-10004-PSOR-008試験(承認年月日:2016年12月19日、CTD2.7.6.20)
- Paul C,et al., Br J Dermatol., 173, 1387-1399, (2015) »PubMed
- CC-10004-PSOR-009試験(承認年月日:2016年12月19日、CTD2.7.6.21)
- Kavanaugh A,et al., Ann Rheum Dis., 73, 1020-1026, (2014) »PubMed »DOI
- Kavanaugh A,et al., J Rheumatol., 42, 479-488, (2015) »PubMed »DOI
- CC-10004-PSA-002試験(承認年月日:2016年12月19日、CTD2.7.6.25)
- CC-10004-PSA-003試験(承認年月日:2016年12月19日、CTD2.7.6.26)
- Edwards CJ,et al., Ann Rheum Dis., 75, 1065-1073, (2016) »PubMed »DOI
- CC-10004-PSA-004試験(承認年月日:2016年12月19日、CTD2.7.6.27)
- CC-10004-PSA-005試験(承認年月日:2016年12月19日、CTD2.7.6.28)
- 20200195試験(承認年月日:2025年3月27日、CTD2.7.6.2)
- CC-10004-BCT-002試験(承認年月日:2019年9月20日、CTD2.7.6.1)
- Schafer PH,et al., Br J Pharmacol., 159 (4), 842-855, (2010) »PubMed
- Schafer PH,et al., Cell Signal., 26 (9), 2016-2029, (2014) »PubMed
- McCann FE,et al., Arthritis Res Ther., 12 (3), R107, (2010) »PubMed
24. 文献請求先及び問い合わせ先
文献請求先
アムジェン株式会社
メディカルインフォメーションセンター
〒107-6239
東京都港区赤坂九丁目7番1号
電話:0120-790-549
製品情報問い合わせ先
アムジェン株式会社
メディカルインフォメーションセンター
〒107-6239
東京都港区赤坂九丁目7番1号
電話:0120-790-549
26. 製造販売業者等
26.1 製造販売元
アムジェン株式会社
東京都港区赤坂九丁目7番1号