医薬品情報
| 総称名 | リリカ |
|---|---|
| 一般名 | プレガバリン |
| 欧文一般名 | Pregabalin |
| 製剤名 | プレガバリンカプセル・プレガバリン口腔内崩壊錠 |
| 薬効分類名 | 疼痛治療剤(神経障害性疼痛・線維筋痛症) |
| 薬効分類番号 | 1190 |
| ATCコード | N02BF02 |
| KEGG DRUG |
D02716
プレガバリン
|
| JAPIC | 添付文書(PDF) |
添付文書情報2025年12月 改訂(第8版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| リリカカプセル25mg | LYRICA Capsules | ヴィアトリス製薬 | 1190017M1028 | 23.7円/カプセル | 処方箋医薬品注) |
| リリカカプセル75mg | LYRICA Capsules | ヴィアトリス製薬 | 1190017M2024 | 38.4円/カプセル | 処方箋医薬品注) |
| リリカカプセル150mg | LYRICA Capsules | ヴィアトリス製薬 | 1190017M3020 | 53.2円/カプセル | 処方箋医薬品注) |
| リリカOD錠25mg | LYRICA OD Tablets | ヴィアトリス製薬 | 1190017F1029 | 23.7円/錠 | 処方箋医薬品注) |
| リリカOD錠75mg | LYRICA OD Tablets | ヴィアトリス製薬 | 1190017F2025 | 38.4円/錠 | 処方箋医薬品注) |
| リリカOD錠150mg | LYRICA OD Tablets | ヴィアトリス製薬 | 1190017F3021 | 53.2円/錠 | 処方箋医薬品注) |
2. 禁忌
次の患者には投与しないこと
本剤の成分に対し過敏症の既往歴のある患者
4. 効能または効果
5. 効能または効果に関連する注意
<線維筋痛症に伴う疼痛>
線維筋痛症の診断は、米国リウマチ学会の分類(診断)基準等の国際的な基準に基づき慎重に実施し、確定診断された場合にのみ投与すること。
6. 用法及び用量
<神経障害性疼痛>
通常、成人には初期用量としてプレガバリン1日150mgを1日2回に分けて経口投与し、その後1週間以上かけて1日用量として300mgまで漸増する。なお、年齢、症状により適宜増減するが、1日最高用量は600mgを超えないこととし、いずれも1日2回に分けて経口投与する。
<線維筋痛症に伴う疼痛>
通常、成人には初期用量としてプレガバリン1日150mgを1日2回に分けて経口投与し、その後1週間以上かけて1日用量として300mgまで漸増した後、300〜450mgで維持する。なお、年齢、症状により適宜増減するが、1日最高用量は450mgを超えないこととし、いずれも1日2回に分けて経口投与する。
7. 用法及び用量に関連する注意
7.1 本剤の投与を中止する場合には、少なくとも1週間以上かけて徐々に減量すること。[8.2参照]
7.2 腎機能障害患者に本剤を投与する場合は、下表に示すクレアチニンクリアランス値を参考として本剤の投与量及び投与間隔を調節すること。また、血液透析を受けている患者では、クレアチニンクリアランス値に応じた1日用量に加えて、血液透析を実施した後に本剤の追加投与を行うこと。複数の用量が設定されている場合には、低用量から開始し、忍容性が確認され、効果不十分な場合に増量すること。なお、ここで示している用法・用量はシミュレーション結果に基づくものであることから、各患者ごとに慎重に観察しながら、用法・用量を調節すること。[9.2、9.8.1、16.6.2参照]
| クレアチニンクリアランス(mL/min) | ≧60 | ≧30-<60 | ≧15-<30 | <15 | 血液透析後の補充用量注) |
| 1日投与量 | 150〜600mg | 75〜300mg | 25〜150mg | 25〜75mg | / |
| 初期用量 | 1回75mg1日2回 | 1回25mg1日3回 又は 1回75mg1日1回 | 1回25mg1日1回もしくは2回 又は 1回50mg1日1回 | 1回25mg1日1回 | 25又は50mg |
| 維持量 | 1回150mg1日2回 | 1回50mg1日3回 又は 1回75mg1日2回 | 1回75mg1日1回 | 1回25又は50mg1日1回 | 50又は75mg |
| 最高投与量 | 1回300mg1日2回 | 1回100mg1日3回 又は 1回150mg1日2回 | 1回75mg1日2回 又は 1回150mg1日1回 | 1回75mg1日1回 | 100又は150mg |
| クレアチニンクリアランス(mL/min) | ≧60 | ≧30-<60 | ≧15-<30 | <15 | 血液透析後の補充用量注) |
| 1日投与量 | 150〜450mg | 75〜225mg | 25〜150mg | 25〜75mg | / |
| 初期用量 | 1回75mg1日2回 | 1回25mg1日3回 又は 1回75mg1日1回 | 1回25mg1日1回もしくは2回 又は 1回50mg1日1回 | 1回25mg1日1回 | 25又は50mg |
| 維持量 | 1回150mg1日2回 | 1回50mg1日3回 又は 1回75mg1日2回 | 1回75mg1日1回 | 1回25又は50mg1日1回 | 50又は75mg |
| 維持量(最高投与量) | 1回225mg1日2回 | 1回75mg1日3回 | 1回100もしくは125mg1日1回 又は 1回75mg1日2回 | 1回50又は75mg1日1回 | 75又は100mg |
8. 重要な基本的注意
<効能共通>
8.1 本剤の投与によりめまい、傾眠、意識消失等があらわれ、自動車事故に至った例もあるので、本剤投与中の患者には、自動車の運転等危険を伴う機械の操作に従事させないよう注意すること。[11.1.1参照]
8.2 本剤の急激な投与中止により、不眠、悪心、頭痛、下痢、不安及び多汗症等の離脱症状があらわれることがあるので、投与を中止する場合には、少なくとも1週間以上かけて徐々に減量すること。[7.1参照]
8.3 本剤の投与により体重増加を来すことがあるので、肥満に注意し、肥満の徴候があらわれた場合は、食事療法、運動療法等の適切な処置を行うこと。特に、投与量の増加、あるいは長期投与に伴い体重増加が認められることがあるため、定期的に体重計測を実施すること。
8.4 本剤の投与により、弱視、視覚異常、霧視、複視等の眼障害が生じる可能性があるので、診察時に、眼障害について問診を行う等注意し、異常が認められた場合には適切な処置を行うこと。[15.2.2参照]
<神経障害性疼痛>
8.5 本剤による神経障害性疼痛の治療は原因療法ではなく対症療法であることから、疼痛の原因となる疾患の診断及び治療を併せて行い、本剤を漫然と投与しないこと。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 重度のうっ血性心不全の患者
心血管障害を有する患者において、うっ血性心不全があらわれることがある。[11.1.2参照]
9.1.2 血管浮腫の既往がある患者[11.1.5参照]
9.1.3 薬物依存の傾向のある患者又は既往歴のある患者、精神障害のある患者
依存の兆候がないかを観察し、慎重に投与すること。[15.1.2参照]
9.2 腎機能障害患者
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。動物実験で、胎児異常(低体重、限局性浮腫の発生率上昇、骨格変異、骨化遅延等)、出生児への影響(体重低下、生存率の低下、聴覚性驚愕反応の低下、発育遅延、生殖能に対する影響等)が報告されている1)。
9.6 授乳婦
9.7 小児等
小児等を対象とした有効性及び安全性を指標とした臨床試験は実施していない。幼若ラットでは本薬の感受性が高く、最大臨床用量(600mg/日)と同等の曝露において、中枢神経症状(自発運動亢進及び歯ぎしり)及び成長への影響(一過性の体重増加抑制)が報告されている。また、最大臨床用量の2倍を超える曝露で聴覚性驚愕反応の低下が、約5倍の曝露で発情休止期の延長が報告されている3)。
9.8 高齢者
10. 相互作用
10.2 併用注意
| 中枢神経抑制剤 オピオイド系鎮痛剤 | 呼吸不全、昏睡がみられたとの報告がある。 | 機序不明 |
| オキシコドン ロラゼパム アルコール(飲酒) | 認知機能障害及び粗大運動機能障害に対して本剤が相加的に作用するおそれがある。 | 相加的な作用による |
| 血管浮腫を引き起こす薬剤(アンジオテンシン変換酵素阻害薬等) | 血管浮腫との関連性が示されている薬剤を服用している患者では、血管浮腫(顔面、口、頸部の腫脹など)を発症するリスクが高まるおそれがある。 | 機序不明 |
| 末梢性浮腫を引き起こす薬剤(チアゾリジン系薬剤等) | チアゾリジン系薬剤と本剤の併用により末梢性浮腫を発症するリスクが高まるおそれがある。また、チアゾリジン系薬剤は体重増加又は体液貯留を引き起こし、心不全が発症又は悪化することがあるため、本剤と併用する場合には慎重に投与すること。 | 機序不明 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 めまい(20%以上)、傾眠(20%以上)、意識消失(0.3%未満)
11.1.2 心不全(0.3%未満)、肺水腫(頻度不明)
心不全のリスクがある患者では、観察を十分に行い、異常が認められた場合には投与を中止し、適切な処置を行うこと。[9.1.1参照]
11.1.3 横紋筋融解症(頻度不明)
筋肉痛、脱力感、CK上昇、血中及び尿中ミオグロビン上昇等があらわれた場合には、投与を中止し、適切な処置を行うこと。また、横紋筋融解症による急性腎障害の発症に注意すること。
11.1.4 腎不全(0.1%未満)
11.1.5 血管浮腫(頻度不明)
血管浮腫等の過敏症があらわれることがある。[9.1.2参照]
11.1.6 低血糖(0.3%未満)
脱力感、倦怠感、冷汗、振戦、意識障害等の低血糖症状があらわれた場合には投与を中止し、適切な処置を行うこと。
11.1.7 間質性肺炎(頻度不明)
咳嗽、呼吸困難、発熱等の臨床症状を十分に観察し、異常が認められた場合には胸部X線、胸部CT等の検査を実施すること。間質性肺炎が疑われた場合には投与を中止し、副腎皮質ホルモン剤の投与等の適切な処置を行うこと。
11.1.8 ショック(頻度不明)、アナフィラキシー(0.1%未満)
11.1.9 皮膚粘膜眼症候群(Stevens-Johnson症候群)(頻度不明)、多形紅斑(頻度不明)
11.1.10 劇症肝炎(頻度不明)、肝機能障害(0.4%)
劇症肝炎、AST、ALT上昇等を伴う肝機能障害があらわれることがある。
11.2 その他の副作用
| 1%以上 | 0.3%以上1%未満 | 0.3%未満 | 頻度不明 | |
| 血液及びリンパ系障害 | 好中球減少症、白血球減少症 | 血小板減少症 | ||
| 代謝及び栄養障害 | 食欲不振、食欲亢進、高脂血症 | 高血糖 | ||
| 精神障害 | 不眠症 | 錯乱、失見当識、多幸気分、異常な夢、幻覚 | うつ病、落ち着きのなさ、気分動揺、抑うつ気分、無感情、不安、リビドー消失、睡眠障害、思考異常 | 離人症、無オルガズム症、激越、喚語困難、リビドー亢進、パニック発作、脱抑制 |
| 神経系障害 | 浮動性めまい、頭痛、平衡障害、運動失調 | 振戦、注意力障害、感覚鈍麻、嗜眠、構語障害、記憶障害、健忘、錯感覚、協調運動異常 | 鎮静、認知障害、ミオクローヌス、反射消失、ジスキネジー、精神運動亢進、体位性めまい、知覚過敏、味覚異常、灼熱感、失神、精神的機能障害、会話障害 | 昏迷、嗅覚錯誤、書字障害 |
| 眼障害 | 霧視、複視、視力低下 | 視覚障害、網膜出血 | 視野欠損、眼部腫脹、眼痛、眼精疲労、流涙増加、光視症、斜視、眼乾燥、眼振 | 眼刺激、散瞳、動揺視、深径覚の変化、視覚の明るさ、角膜炎 |
| 耳及び迷路障害 | 回転性めまい | 耳鳴 | 聴覚過敏 | |
| 心臓障害 | 動悸 | 第一度房室ブロック、頻脈、洞性不整脈、洞性徐脈、心室性期外収縮 | 洞性頻脈 | |
| 血管障害 | 高血圧、低血圧、ほてり | |||
| 呼吸器、胸郭及び縦隔障害 | 呼吸困難 | 鼻咽頭炎、咳嗽、いびき、鼻出血、鼻炎 | 鼻乾燥、鼻閉、咽喉絞扼感 | |
| 胃腸障害 | 便秘、悪心、下痢、腹痛、嘔吐 | 腹部膨満、消化不良、鼓腸、胃炎、胃不快感、口内炎 | 流涎過多、胃食道逆流性疾患、膵炎、舌腫脹 | 腹水、嚥下障害 |
| 皮膚及び皮下組織障害 | 発疹 | そう痒症、湿疹、眼窩周囲浮腫 | 多汗症、冷汗、蕁麻疹、脱毛 | 丘疹 |
| 筋骨格系及び結合組織障害 | 筋力低下、筋痙縮、関節腫脹、四肢痛、背部痛 | 筋肉痛、重感、関節痛、筋骨格硬直 | ||
| 腎及び尿路障害 | 尿失禁、排尿困難 | 尿閉 | 乏尿 | |
| 生殖系及び乳房障害 | 乳房痛、勃起不全、女性化乳房 | 射精遅延、性機能不全、無月経、乳房分泌、月経困難症、乳房肥大 | ||
| 全身障害及び投与局所様態 | 浮腫、口渇、疲労、異常感、歩行障害、顔面浮腫 | 無力症、疼痛、圧痕浮腫、倦怠感、胸痛 | 発熱、冷感、悪寒、易刺激性、酩酊感 | 胸部絞扼感 |
| 傷害、中毒及び処置合併症 | 転倒・転落 | |||
| 臨床検査 | 体重増加 | 血中CK増加、ALT増加、AST増加、血中アミラーゼ増加、血中クレアチニン増加 | 体重減少、血中尿酸増加 | 血中カリウム減少 |
13. 過量投与
13.1 症状
15gまでの過量投与例が報告されており、過量投与時にみられた主な症状は、情動障害、傾眠、錯乱状態、抑うつ、激越、落ち着きのなさ、痙攣発作である。
13.2 処置
本剤は血液透析により除去されることから、発現している症状の程度に応じて血液透析の実施を考慮すること。[16.6.3参照]
14. 適用上の注意
14.1 薬剤交付時の注意
<製剤共通>
14.1.1 PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
<OD錠>
14.1.2 本剤は舌の上にのせて唾液を湿潤させると崩壊するため、水なしで服用可能である。また、水で服用することもできる。
15. その他の注意
15.1 臨床使用に基づく情報
15.1.1 海外で実施された本剤を含む複数の抗てんかん薬における、てんかん、精神疾患等を対象とした199のプラセボ対照臨床試験の検討結果において、自殺念慮及び自殺企図の発現のリスクが、抗てんかん薬の服用群でプラセボ群と比較して約2倍高く(抗てんかん薬服用群:0.43%、プラセボ群:0.24%)、抗てんかん薬の服用群では、プラセボ群と比べ1000人あたり1.9人多いと計算された(95%信頼区間:0.6-3.9)。また、てんかん患者のサブグループでは、プラセボ群と比べ1000人あたり2.4人多いと計算されている注)。
注)本剤は海外で抗てんかん薬として承認されているが、本邦における本剤の効能・効果は「神経障害性疼痛、線維筋痛症に伴う疼痛」である。
15.1.2 薬物乱用に関連する受容体部位の活性作用は知られていないが、本剤を投与された患者で依存の症例が市販後に報告されている。[9.1.3参照]
15.2 非臨床試験に基づく情報
15.2.1 2年間のマウスがん原性試験において、最大臨床用量での平均ヒト曝露量の6倍以上の曝露量に相当する本薬の投与により、用量依存的に血管肉腫の発生率が増加したとの報告がある4)。
15.2.2 2年間のラットがん原性試験において、最大臨床用量での平均ヒト曝露量の5倍以上の曝露量に相当する本薬の投与により、加齢アルビノラットに通常認められる網膜萎縮の発現率が増加したとの報告がある4)。また、ラットを用いた組織分布試験において、水晶体での14C-プレガバリン由来放射能の消失は血液及びほとんどの組織にくらべ緩徐であったが、ラット13及び52週間反復投与毒性試験では水晶体に対する影響は認められなかった。眼に関する副作用の発現率はプラセボ群より高く、神経障害性疼痛を対象とした13〜16週間投与のプラセボ対照試験(3試験併合)のプラセボ群では3.8%に対し、本剤群(150〜600mg/日)で10.6%、長期投与試験(3試験併合)では10.2%、線維筋痛症を対象とした16週間投与のプラセボ対照試験のプラセボ群では2.8%に対し、本剤群(300〜450mg/日)で9.2%、長期投与試験では9.4%であった。[8.4参照]
15.2.3 雄ラットの受胎能及び初期胚発生に関する試験において、最大臨床用量での平均ヒト曝露量の28倍以上の曝露量に相当する本薬の投与により、胎児異常の発生頻度が増加したとの報告がある1)。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
日本人健康成人に、プレガバリン50、100、200、250及び300mg(各投与量6例)を絶食時に単回経口投与した時、投与後約1時間でCmaxに達し、T1/2は約6時間であった。Cmax及びAUC0-∞は、300mgまでの用量範囲で、用量に比例して増加した5)。
| 投与量(mg) | Cmax(μg/mL) | Tmax(h) | AUC0-∞(μg・h/mL) | T1/2(h) | CL/F(L/h) | Vd/F(L) | Ae(%) |
| 50 | 2.03(0.40) | 0.67(0.26) | 10.7(1.1) | 5.98(0.65) | 4.72(0.44) | 40.6(4.9) | 83.9(5.4) |
| 100 | 3.56(0.67) | 0.75(0.27) | 20.4(1.3) | 5.66(0.59) | 4.93(0.35) | 40.3(6.4) | 95.0(2.7) |
| 200 | 6.35(0.73) | 1.00(0.32) | 43.2(3.0) | 5.93(0.32) | 4.64(0.32) | 39.7(2.7) | 91.8(2.6) |
| 250 | 7.18(1.43) | 1.17(0.52) | 49.2(6.1) | 5.57(0.72) | 5.15(0.61) | 41.0(3.8) | 95.6(4.4) |
| 300 | 8.25(1.36) | 1.08(0.38) | 61.7(6.3) | 5.80(0.62) | 4.91(0.52) | 40.9(4.3) | 97.7(7.3) |
16.1.2 反復投与
日本人健康成人にプレガバリン1回150及び300mg(各投与量7例)を1日2回7日間反復経口投与した時、投与後24〜48時間で定常状態に達し、投与7日目のT1/2はそれぞれ5.95及び6.31時間であった。投与7日目のAUC0-12は、投与第1日目の1.4倍であった6)。
| Cmax(μg/mL) | Tmax(h) | AUC0-12(μg・h/mL) | T1/2(h) | |||||
| 第1日 | 第7日 | 第1日 | 第7日 | 第1日 | 第7日 | 第1日 | 第7日 | |
| 1回150mg (1日2回) | 4.36(0.68) | 6.24(0.79) | 0.9(0.4) | 0.9(0.5) | 21.8(1.7) | 30.7(2.9) | 5.11(0.75) | 5.95(0.46) |
| 1回300mg (1日2回) | 8.73(2.52) | 10.5(2.4) | 1.6(1.1) | 1.6(0.8) | 41.9(7.4) | 58.7(11.4) | 5.40(0.94) | 6.31(0.58) |
16.1.3 生物学的同等性
日本人健康成人24例に、クロスオーバー法によりプレガバリンOD錠150mg又はプレガバリンカプセル150mgをそれぞれ空腹時単回経口投与したとき、プレガバリンの血漿中濃度及び薬物動態パラメータは以下のとおりであった。OD錠150mgは、水なし又は水で服用した場合のいずれにおいても、プレガバリンカプセル150mgと生物学的に同等であることが確認された7)。
| 剤形及び投与量 | Cmax(μg/mL) | AUCt(μg・h/mL) | Tmax(h) | T1/2(h) |
| OD錠150mg (水なしで服用) | 5.794(1.180) | 31.180(4.958) | 1.00(0.333-2.50) | 5.955(0.679) |
| OD錠150mg (水で服用) | 5.793(1.421) | 31.410(4.560) | 0.500(0.333-1.50) | 6.008(0.698) |
| プレガバリンカプセル150mg (水で服用) | 5.787(1.231) | 31.690(4.477) | 0.875(0.500-2.00) | 6.036(0.698) |
また、OD錠150mgはOD錠25mg及びOD錠75mgと溶出挙動が同等であり、生物学的に同等とみなされた。
16.2 吸収
16.2.1 食事の影響
日本人健康成人19例において、絶食時及び食後にプレガバリンを150mg単回経口投与した時のCmaxはそれぞれ4.95及び3.22μg/mL、Tmaxは0.947及び3.37時間、AUC0-48はそれぞれ31.2及び28.8μg・h/mLであった。食後投与においてCmaxは約35%低下し、Tmaxは約2.4時間延長したが、AUC0-48の低下は約8%であった8)。
16.3 分布
16.4 代謝
16.5 排泄
日本人健康成人に、プレガバリン50、100、200、250及び300mg(各投与量6例)を絶食時に単回経口投与した時のCL/Fは4.64〜5.15L/hであった。この時の尿中排泄率は83.9〜97.7%であった5)。
16.6 特定の背景を有する患者
16.6.1 高齢者
年齢が67〜78歳の日本人健康高齢者6例にプレガバリン100mgを単回経口投与した時、Tmaxは1.4時間、T1/2は6.32時間であった。AUC0-∞及びT1/2は、健康非高齢者にプレガバリン100mgを単回経口投与した時と比較してわずかに増大及び延長する傾向が確認された12)。
| Cmax(μg/mL) | Tmax(h) | AUC0-∞(μg・h/mL) | T1/2(h) | CL/F(L/h) | |
| 健康高齢者 | 3.24(0.55) | 1.4(0.5) | 26.6(4.3) | 6.32(0.82) | 3.82(0.65) |
| 健康非高齢者 | 3.56(0.67) | 0.75(0.27) | 20.4(1.3) | 5.66(0.59) | 4.93(0.35) |
16.6.2 腎機能障害患者
(1)腎機能の異なる被験者26例を対象に、プレガバリン50mgを単回経口投与した時、腎機能の低下に従ってT1/2が延長し、AUC0-∞が増加した。CL/F及び腎クリアランス(CLr)はクレアチニンクリアランスに比例した13)(外国人データ)。
| クレアチニンクリアランス | Cmax(μg/mL) | Tmax(h) | AUC0-∞(μg・h/mL) | T1/2(h) | CL/F(mL/min) | CLr(mL/min) |
| ≧60mL/min (n=11) | 1.86(0.39) | 1.00(0.224) | 15.9(4.4) | 9.11(2.83) | 56.5(17.6) | 44.9(23.6) |
| ≧30-<60mL/min (n=7) | 1.53(0.29) | 1.29(0.393) | 28.2(5.0) | 16.7(4.1) | 30.6(7.3) | 15.4(7.7) |
| ≧15-<30mL/min (n=7) | 1.90(0.62) | 1.93(1.48) | 52.3(11.7) | 25.0(6.7) | 16.7(3.9) | 9.23(3.37) |
| <15mL/min (n=1) | 1.69 | 1.00 | 101 | 48.7 | 8.30 | 4.30 |
(2)母集団薬物動態解析
838例の被験者(日本人474例を含む:健康被験者70例、帯状疱疹後神経痛患者26例、糖尿病性末梢神経障害に伴う疼痛を有する患者154例及び線維筋痛症患者224例)を対象として母集団薬物動態解析を実施した結果、一次吸収を含む1-コンパートメントモデルが構築され、共変量としてCL/Fに対してクレアチニンクリアランス(CLcr)及び理想体重、Vd/Fに対してBMI、理想体重、性別及び年齢が同定されたが、プレガバリンの薬物動態に影響を与える因子としてはCL/Fに対するCLcrが重要であると考えられた。腎機能障害患者において、CLcrの低下により、プレガバリンのCL/Fは低下するため、CLcr値を参考とした用法・用量の調節が必要である。
また、日本人の糖尿病性末梢神経障害に伴う疼痛患者において、CLcrが30mL/min以上60mL/min未満に低下している患者にプレガバリン150mgを1日2回反復経口投与(300mg/日)したときの定常状態におけるAUC0-12(AUC0-12,ss)のモデルによる推定値は、CLcrが60mL/min以上の患者にプレガバリン300mgを1日2回反復経口投与(600mg/日)したときと同じであった。CLcrが30mL/min以上60mL/min未満の患者におけるプレガバリンのクリアランスは、CLcrが60mL/min以上の患者の約半分であった14)。[7.2、9.2、9.8.1参照]
また、日本人の糖尿病性末梢神経障害に伴う疼痛患者において、CLcrが30mL/min以上60mL/min未満に低下している患者にプレガバリン150mgを1日2回反復経口投与(300mg/日)したときの定常状態におけるAUC0-12(AUC0-12,ss)のモデルによる推定値は、CLcrが60mL/min以上の患者にプレガバリン300mgを1日2回反復経口投与(600mg/日)したときと同じであった。CLcrが30mL/min以上60mL/min未満の患者におけるプレガバリンのクリアランスは、CLcrが60mL/min以上の患者の約半分であった14)。[7.2、9.2、9.8.1参照]
| クレアチニンクリアランス | 投与量 | AUC0-12,ss(μg・h/mL) | CL/F(mL/min) |
| ≧60mL/min (n=31) | 1回300mg (1日2回) | 86.1(27.8) | 63.6(18.5) |
| ≧30-<60mL/min (n=14) | 1回150mg (1日2回) | 85.7(22.6) | 31.1(8.11) |
16.6.3 血液透析患者
16.6.4 授乳婦
16.7 薬物相互作用
本剤は主として未変化体のまま尿中に排泄され、ヒトにおいてほとんど代謝されることなく、また血漿蛋白にも結合しないため、本剤が薬物相互作用を引き起こす可能性は低い(外国人データ)。
16.7.1 ガバペンチン
本剤とガバペンチンの薬物相互作用について、健康成人11例を対象にプレガバリン100mg及びガバペンチン300mgを単回投与した試験、及び健康成人18例にプレガバリン100mg及びガバペンチン400mgを反復投与(投与間隔:8時間)した試験を実施して検討した。その結果、単回投与及び反復投与のいずれにおいても、本剤の併用によってガバペンチンの薬物動態は変化しなかった。また、プレガバリンの吸収速度はガバペンチン併用によってわずかに低下したが、吸収量には影響がなかった15)。
16.7.2 経口避妊薬(酢酸ノルエチンドロン及びエチニルエストラジオールの合剤)
健康成人女性16例を対象に経口避妊薬(酢酸ノルエチンドロン1mg及びエチニルエストラジオール0.035mgの合剤1日1回)とプレガバリン(1回200mg1日3回)を同時に経口投与した時、プレガバリン併用時のノルエチンドロンのCmaxはプレガバリン非併用時と比較して変化せず、プレガバリン併用時のAUC0-24はプレガバリン非併用時と比較して16%増加し、プレガバリンはノルエチンドロンの薬物動態に影響を及ぼさなかった。プレガバリン併用時のエチニルエストラジオールのCmax及びAUC0-24は、プレガバリン非併用時と比較してそれぞれ5%及び14%増加し、プレガバリンはエチニルエストラジオールの薬物動態に影響を及ぼさなかった。また、経口避妊薬はプレガバリンの血漿中濃度(トラフ値)に影響を及ぼさなかった16)。
16.7.3 ロラゼパム
健康成人12例を対象にプレガバリン(1回300mg1日2回)を反復経口投与後、ロラゼパム(1mg)を併用投与した時、ロラゼパムのCmax及びAUC0-∞は、プレガバリン非併用時と比較してそれぞれ6%及び8%増加し、プレガバリンはロラゼパムの薬物動態に影響を及ぼさなかった。また、ロラゼパム併用時のプレガバリンのCmaxは、ロラゼパム非併用時より2%増加し、AUC0-12は1.8%低く、ロラゼパムはプレガバリンの薬物動態に影響を与えなかった。プレガバリンとロラゼパムの併用により、認知機能及び粗大運動機能における反応速度や正答率等が、単剤投与時に比べて相加的に低下する傾向が認められた17)。
16.7.4 オキシコドン
健康成人12例を対象にプレガバリン(1回300mg1日2回)を反復経口投与後、オキシコドン(10mg)を併用投与した時、オキシコドンのCmax及びAUC0-∞は、プレガバリン非併用時と比較してそれぞれ1.1%及び9.5%減少し、プレガバリンはオキシコドンの薬物動態に影響を及ぼさなかった。また、オキシコドン併用時のプレガバリンのCmaxは、オキシコドン非併用時より4.5%低かったが、AUC0-12は同程度であり、オキシコドンはプレガバリンの薬物動態に影響を与えなかった。プレガバリンとオキシコドンの併用により、認知機能及び粗大運動機能における反応速度や正答率等が、単剤投与時に比べて相加的に低下する傾向が認められた18)。
16.7.5 エタノール
健康成人13例を対象にプレガバリン(1回300mg1日2回)を反復経口投与後、エタノール(0.70g/kg)を併用投与した時、エタノールのCmax及びAUC0-∞は、プレガバリン非併用時と比較してそれぞれ8.9%及び9.6%減少し、プレガバリンはエタノールの薬物動態に影響を及ぼさなかった。また、エタノール併用時のプレガバリンのCmax及びAUC0-12は、エタノール非併用時と比較してそれぞれ21%及び1%高かったが、この差は臨床上問題となる差ではないと考えられた。プレガバリンとエタノールの併用により、認知機能及び粗大運動機能における反応速度や正答率等が、単剤投与時に比べて相加的に低下する傾向が認められた19)。
16.7.6 フェニトイン
フェニトイン単剤の維持投与により症状が安定している成人部分てんかん患者10例を対象にプレガバリン(1回200mg1日3回)を反復経口投与した時、プレガバリンはフェニトインの血漿中濃度(トラフ値)に影響を及ぼさず、またフェニトインもプレガバリンの薬物動態に影響を与えなかった20)。
16.7.7 カルバマゼピン
カルバマゼピン単剤を維持投与されている成人てんかん患者12例を対象にプレガバリン(1回200mg1日3回)を反復経口投与した時、プレガバリンはカルバマゼピン及びその代謝物(10,11-エポキシド体)の血漿中濃度(トラフ値)に影響を及ぼさず、またカルバマゼピンもプレガバリンの薬物動態に影響を与えなかった20)。
16.7.8 バルプロ酸
バルプロ酸ナトリウム単剤を維持投与されている成人てんかん患者12例を対象にプレガバリン(1回200mg1日3回)を反復経口投与した時、プレガバリンはバルプロ酸の血漿中濃度(トラフ値)に影響を及ぼさず、またバルプロ酸もプレガバリンの薬物動態に影響を与えなかった20)。
16.7.9 ラモトリギン
ラモトリギンを単剤で維持投与されている成人てんかん患者12例を対象にプレガバリン(1回200mg1日3回)を反復経口投与した時、プレガバリンはラモトリギンの血漿中濃度(トラフ値)に影響を及ぼさず、またラモトリギンもプレガバリンの薬物動態に影響を与えなかった20)。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国内プラセボ対照試験
帯状疱疹後神経痛患者を対象とした13週間投与の二重盲検比較試験において、主要評価項目である最終評価時の疼痛スコアは下表のとおりであり、プレガバリン300mg/日群及び600mg/日群ではプラセボ群と比較して統計学的に有意な改善が認められた21)。
| 投与群 | 最終評価時の疼痛スコアa,b) | |||
| 症例数 | 最小二乗平均±標準誤差 | プラセボとの差[95%信頼区間] | p値 | |
| プラセボ | 97 | 5.12±0.19 | − | − |
| プレガバリン150mg/日 | 86 | 4.81±0.20 | −0.31[−0.85,0.23] | 0.262 |
| プレガバリン300mg/日 | 89 | 4.26±0.20 | −0.86[−1.39,−0.32] | 0.002 |
| プレガバリン600mg/日c) | 97 | 4.49±0.19 | −0.63[−1.15,−0.10] | 0.019 |
安全性評価対象例中の副作用発現率は、プラセボ群で43.9%(43/98例)、プレガバリン150mg/日群で57.5%(50/87例)、300mg/日群で73.0%(65/89例)、600mg/日群で82.5%(80/97例)であった。主な副作用は、浮動性めまい(31.1%)、傾眠(28.6%)、便秘(12.1%)、末梢性浮腫(11.7%)であった。重篤な副作用は、150mg/日群に心筋梗塞1例(転帰:未回復)、300mg/日群に意識消失/低血圧の1例2件(転帰:回復)が報告された。
糖尿病性末梢神経障害に伴う疼痛患者を対象とした13週間投与の二重盲検比較試験において、主要評価項目である最終評価時の疼痛スコアは下表のとおりであり、プレガバリン300mg/日群ではプラセボ群と比較して統計学的に有意な改善が認められた22)。
| 投与群 | 最終評価時の疼痛スコアd,e) | ||||
| 症例数 | 最小二乗平均±標準誤差 | ベースラインからの変化量最小二乗平均±標準誤差 | プラセボとの差[95%信頼区間] | p値 | |
| プラセボ | 135 | 4.83±0.21 | −1.20±0.21 | − | − |
| プレガバリン300mg/日 | 134 | 4.20±0.22 | −1.82±0.22 | −0.63[−1.09,−0.17] | 0.0075 |
| プレガバリン600mg/日f) | 45 | 4.08±0.32 | −1.94±0.32 | −0.74[−1.39,−0.09] | − |
安全性評価対象例中の副作用発現率は、プラセボ群で36.3%(49/135例)、プレガバリン300mg/日群で56.7%(76/134例)、600mg/日群で80.0%(36/45例)であった。主な副作用は、浮動性めまい(300mg/日群19.4%及び600mg/日群37.8%)、傾眠(300mg/日群20.9%及び600mg/日群40.0%)、末梢性浮腫(300mg/日群12.7%及び600mg/日群13.3%)、体重増加(300mg/日群11.2%及び600mg/日群11.1%)であった。
線維筋痛症患者を対象とした16週間投与の二重盲検比較試験において、主要評価項目である最終評価時の疼痛スコアは下表のとおりであり、プレガバリン群(300〜450mg/日)でプラセボ群と比較して統計学的に有意な改善が認められた23)。
| 投与群 | 最終評価時の疼痛スコアg,h) | |||
| 症例数 | 最小二乗平均±標準誤差 | プラセボとの差[95%信頼区間] | p値 | |
| プラセボ | 248 | 5.45±0.12 | − | − |
| プレガバリン300-450mg/日 | 250 | 5.01±0.12 | −0.44[−0.78,−0.11] | 0.0046 |
安全性評価対象例中の副作用発現率は、プラセボ群で51.6%(128/248例)、プレガバリン群で82.4%(206/250例)であった。主な副作用は、傾眠(45.2%)、浮動性めまい(28.8%)、体重増加(14.4%)、便秘(12.8%)であった。
17.1.2 国内長期投与試験
帯状疱疹後神経痛患者126例、糖尿病性末梢神経障害に伴う疼痛患者123例、線維筋痛症患者106例又は中枢性神経障害性疼痛(脊髄損傷後疼痛、脳卒中後疼痛、多発性硬化症に伴う疼痛)患者103例を対象とした長期投与試験(いずれも52週)における痛みの強度(0〜100mmのVisual Analog Scaleで値が大きいほど強い痛みを示す)の平均値は下表のとおりであった24)25)26)27)。
| 評価時点 | 痛みの強度(mm)a) | |||
| 帯状疱疹後神経痛 | 糖尿病性末梢神経障害に伴う疼痛 | |||
| 評価例数 | 平均値±標準偏差 | 評価例数 | 平均値±標準偏差 | |
| 投与前 | 126 | 62.0±19.0 | 123 | 52.8±21.7 |
| 12週 | 116 | 35.3±22.3 | 119 | 30.0±23.0 |
| 24週 | 105 | 34.0±23.0 | 112 | 27.7±22.0 |
| 52週 | 94 | 28.3±22.9 | 97 | 24.8±20.8 |
| 評価時点 | 痛みの強度(mm)a) | |||
| 線維筋痛症 | 中枢性神経障害性疼痛 | |||
| 評価例数 | 平均値±標準偏差 | 評価例数 | 平均値±標準偏差 | |
| 投与前 | 106 | 61.8±23.5 | 103 | 67.1±16.6 |
| 12週 | 104 | 48.9±23.3 | 98 | 44.3±26.9 |
| 28週 | 101 | 48.3±23.8 | 92 | 46.3±27.1 |
| 52週 | 87 | 47.1±24.8 | 85 | 44.9±27.0 |
a)0〜100mmのVisual Analog Scaleで値が大きいほど強い痛みを示す。
国内長期投与試験(帯状疱疹後神経痛)において、安全性評価対象例中の副作用は78.6%(99/126例)に認められ、主な副作用は浮動性めまい(28.6%)、末梢性浮腫(16.7%)、傾眠(15.1%)、体重増加(13.5%)であった。
国内長期投与試験(糖尿病性末梢神経障害に伴う疼痛)において、安全性評価対象例中の副作用は70.7%(87/123例)に認められ、主な副作用は、傾眠(22.8%)、体重増加(22.0%)、浮動性めまい(20.3%)であった。
国内長期投与試験(線維筋痛症)において、安全性評価対象例中に副作用は84.0%(89/106例)に認められ、主な副作用は傾眠(26.4%)、浮動性めまい(24.5%)、体重増加(18.9%)、便秘(16.0%)であった。副作用の重症度は、多くが軽度であり、重度の副作用は認められなかった。
国内長期投与試験(中枢性神経障害性疼痛)において、先行する国際共同臨床試験から重症度が悪化したあるいは本治験期間中に新たに発現した副作用は、87.4%(90/103例)に認められ、主な副作用は、傾眠(48.5%)、体重増加(28.2%)、浮動性めまい(22.3%)、末梢性浮腫(17.5%)であった。
17.1.3 国際共同臨床試験成績
脊髄損傷後疼痛患者を対象とした16週間投与の二重盲検比較試験において、主要評価項目である治療期の疼痛スコアは下表のとおりであり、プレガバリン群(150〜600mg/日)でプラセボ群と比較して統計学的に有意な改善が認められた28)。
| 投与群a) | 疼痛スコアb,c) | |||
| 症例数 | 治療期のベースラインからの平均変化量最小二乗平均±標準誤差 | プラセボとの差[95%信頼区間] | p値 | |
| プラセボ | 106 | −1.07±0.149 | − | − |
| プレガバリン150-600mg/日 | 105 | −1.66±0.157 | −0.59[−0.98,−0.20] | 0.0032 |
安全性評価対象例中の副作用発現率はプラセボ群で46.7%(50/107例)、プレガバリン群で67.0%(75/112例)であった。主な副作用は、傾眠(33.0%)、浮動性めまい(19.6%)、末梢性浮腫(13.4%)であった。重篤な副作用は、低血糖症1例が報告され、回復した。
17.1.4 外国プラセボ対照試験
帯状疱疹後神経痛患者を対象とした二重盲検比較試験において、主要評価項目である最終評価時の疼痛スコアは下表のとおりであり、いずれの試験においてもプレガバリン300mg/日群及び600mg/日群ではプラセボ群と比較して統計学的に有意な改善が認められた29)。
| 臨床試験(評価期間) | 投与群 | 最終評価時の疼痛スコアa,b) | |||
| 症例数 | 最小二乗平均±標準誤差 | プラセボとの差[95%信頼区間] | p値 | ||
| 外国用量反応試験 | |||||
| 1(13週間) | プラセボ | 93 | 6.14±0.23 | − | − |
| プレガバリン150mg/日 | 87 | 5.26±0.24 | −0.88[−1.53,−0.23] | 0.0077 | |
| プレガバリン300mg/日 | 98 | 5.07±0.23 | −1.07[−1.70,−0.45] | 0.0016 | |
| プレガバリン600mg/日c) | 88 | 4.35±0.24 | −1.79[−2.43,−1.15] | 0.0003 | |
| 外国第II/III相試験 | |||||
| 2(5週間) | プラセボ | 87 | 5.59±0.21 | − | − |
| プレガバリン75mg/日 | 83 | 5.46±0.21 | −0.14[−0.71,0.43] | 0.7999 | |
| プレガバリン150mg/日 | 82 | 5.52±0.22 | −0.07[−0.64,0.50] | 0.7999 | |
| 3(8週間) | プラセボ | 81 | 6.33±0.22 | − | − |
| プレガバリン150mg/日 | 81 | 5.14±0.22 | −1.20[−1.81,−0.58] | 0.0002 | |
| プレガバリン300mg/日 | 76 | 4.76±0.23 | −1.57[−2.20,−0.95] | 0.0002 | |
| 4(8週間) | プラセボ | 84 | 5.29±0.24 | − | − |
| プレガバリン600mg/日c) | 88 | 3.60±0.24 | −1.69[−2.33,−1.05] | 0.0001 | |
外国用量反応試験1において、安全性評価対象例中の副作用発現率はプラセボ群で39.8%(37/93例)、プレガバリン150mg/日群で59.8%(52/87例)、300mg/日群で64.3%(63/98例)、600mg/日群で74.4%(67/90例)であった。主な副作用は、浮動性めまい(150mg/日群16.1%、300mg/日群32.7%及び600mg/日群36.7%)、傾眠(150mg/日群9.2%、300mg/日群11.2%及び600mg/日群25.6%)、末梢性浮腫(150mg/日群12.6%、300mg/日群14.3%及び600mg/日群13.3%)であった。重篤な副作用は、300mg/日群にアナフィラキシー様反応1例、600mg/日群に浮動性めまい/顔面浮腫/筋無力症/末梢性浮腫/傾眠の5件が1例中に報告され、いずれも回復した。
外国第II/III相試験2において、安全性評価対象例中の副作用発現率はプラセボ群で25.0%(22/88例)、プレガバリン75mg/日群で32.1%(27/84例)、150mg/日群で41.0%(34/83例)であった。主な副作用は浮動性めまい(75mg/日群8.3%及び150mg/日群14.5%)及び傾眠(75mg/日群7.1%及び150mg/日群9.6%)、口内乾燥(75mg/日群7.1%及び150mg/日群4.8%)、弱視(75mg/日群1.2%及び150mg/日群8.4%)等であった。
外国第II/III相試験3において、安全性評価対象例中の副作用発現率はプラセボ群で39.5%(32/81例)、プレガバリン150mg/日群で50.6%(41/81例)、300mg/日群で67.1%(51/76例)であった。主な副作用は浮動性めまい(150mg/日群12.3%及び300mg/日群26.3%)、傾眠(150mg/日群14.8%及び300mg/日群23.7%)、口内乾燥(150mg/日群9.9%及び300mg/日群6.6%)等であった。重篤な副作用は、150mg/日群に錯乱、心室性期外収縮、心房性不整脈/心室性期外収縮の3例4件が報告された。これらの転帰について、心室性期外収縮、心房性不整脈/心室性期外収縮は未回復、錯乱は回復が確認された。
外国第II/III相試験4において、安全性評価対象例中の副作用発現率はプラセボ群で36.9%(31/84例)、プレガバリン群で73.0%(65/89例)に認められ、主な副作用は浮動性めまい(27.0%)、傾眠(23.6%)、末梢性浮腫(13.5%)、口内乾燥(10.1%)、弱視(9.0%)及び異常歩行(7.9%)等であった。
17.1.5 外国長期投与試験
帯状疱疹後神経痛患者を対象とした長期投与試験1(154例、最長312週投与)及び2(275例、最長172週投与)における痛みの強度の平均値は下表のとおりであった30)。
| 評価時点 | 痛みの強度(mm)a) | |||
| 長期投与試験1 | 長期投与試験2 | |||
| 評価例数 | 平均値±標準偏差 | 評価例数 | 平均値±標準偏差 | |
| 投与前 | 154 | 69.4±18.7 | 275 | 67.3±17.9 |
| 12週 | 114 | 42.8±26.1 | 211 | 40.3±25.4 |
| 24週 | 91 | 40.5±25.1 | 173 | 41.4±24.9 |
| 52週 | 63 | 38.3±24.6 | 122 | 35.7±24.2 |
| 104週 | 32 | 36.8±23.2 | 78 | 32.5±24.1 |
長期投与試験1において、安全性評価対象例中の副作用は75.3%(116/154例)に認められ、主な副作用は浮動性めまい(20.8%)、体重増加(14.9%)、傾眠(13.6%)、事故による外傷(9.7%)、口内乾燥(8.4%)、末梢性浮腫及び失調(各7.1%)、無力症及び悪心(各6.5%)等であった。重篤な副作用は、事故による外傷3例、上室性頻脈、運動障害、低ナトリウム血症、上室性期外収縮/心室性期外収縮、失神/事故による外傷/事故による外傷5例8件が報告され、いずれも回復した。
長期投与試験2において、安全性評価対象例中の副作用は65.1%(179/275例)に認められ、主な副作用は浮動性めまい(16.0%)、末梢性浮腫(12.7%)、傾眠(10.5%)、無力症、弱視、体重増加及び頭痛(各5.8%)、悪心(5.5%)等であった。重篤な副作用は、肝細胞障害、尿路感染/失神の2例3件が報告され、その転帰はそれぞれ未回復、回復であった。
17.3 その他
17.3.1 食事の影響試験
日本人健康成人を対象として絶食時及び食後にプレガバリンを150mg単回経口投与した時の浮動性めまいの発現率は、食後投与5.3%(1/19例)と比べ絶食時投与30.8%(12/39例)で高かった。
18. 薬効薬理
18.1 作用機序
18.2 鎮痛作用
プレガバリンは、動物実験において急性侵害刺激に対する逃避行動は妨げず、末梢神経損傷及び糖尿病による神経障害性疼痛並びに慢性筋骨格系疼痛を抑制する。また、化学性、炎症性、組織損傷性に惹起される自発痛、痛覚過敏モデルにおいても鎮痛作用を示す36)37)38)39)40)41)。
18.2.1 慢性絞扼神経損傷(CCI)モデルにおける抗アロディニア注)作用
プレガバリンは、ラットCCIモデルによる、静的及び動的アロディニアをともに抑制した36)。
18.2.2 脊髄神経結紮(SNL)モデルにおける抗アロディニア作用
プレガバリンは、SNLモデルによりラットに発生させた静的及び動的アロディニアを抑制した36)。
18.2.3 ストレプトゾシン(STZ)糖尿病モデルにおける抗アロディニア作用
プレガバリンは、ラットSTZ糖尿病モデルにおいて発生する静的及び動的アロディニアを抑制した37)。
18.2.4 脊髄損傷後疼痛モデルにおける抗アロディニア作用
プレガバリンは、マウス脊髄への錘落下による脊髄損傷モデルにおいて発生する静的アロディニアを抑制した38)。
18.2.5 慢性筋骨格系疼痛モデルにおける抗アロディニア作用
プレガバリンは、ラット慢性筋骨格系疼痛モデルにおいて発生する静的アロディニアを抑制した39)。
18.2.6 ホルマリンテストにおける自発痛に対する鎮痛作用
ラット足蹠へのホルマリン投与により発生する2相性の疼痛関連行動のうち、プレガバリンは中枢性感作が関与するとされる第2相を抑制した40)。
注)通常では無害な触覚刺激に対し感じる痛みを接触性アロディニアと呼び、静的(皮膚を軽く点状に圧することで生じる)及び動的(皮膚への軽擦で生じる)アロディニアに分類される。
19. 有効成分に関する理化学的知見
19.1. プレガバリン
| 一般的名称 | プレガバリン |
|---|---|
| 一般的名称(欧名) | Pregabalin |
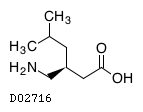
| 化学名 | (3S)-3-(Aminomethyl)-5-methylhexanoic acid |
| 分子式 | C8H17NO2 |
| 分子量 | 159.23 |
| 物理化学的性状 | 白色の粉末である。 水にやや溶けにくく、メタノール又はエタノール(99.5)に溶けにくい。 |
| KEGG DRUG |
|
22. 包装
<リリカカプセル25mg>
100カプセル[10カプセル(PTP)×10]
500カプセル[10カプセル(PTP)×50]
<リリカカプセル75mg>
100カプセル[10カプセル(PTP)×10]
<リリカカプセル150mg>
100カプセル[10カプセル(PTP)×10]
<リリカOD錠25mg>
100錠[10錠(PTP)×10]
500錠[10錠(PTP)×50]
<リリカOD錠75mg>
100錠[10錠(PTP)×10]
500錠[10錠(PTP)×50]
<リリカOD錠150mg>
100錠[10錠(PTP)×10]
23. 主要文献
- 社内資料:生殖発生毒性試験(2010年4月16日承認、CTD2.6.6.6)
- 社内資料:健康授乳婦における薬物動態
- 社内資料:幼若動物を用いた毒性試験(2010年4月16日承認、CTD2.6.6.6)
- 社内資料:がん原性試験(2010年4月16日承認、CTD2.6.6.5)
- 社内資料:健康成人における薬物動態(単回投与)(2010年4月16日承認、CTD2.7.2.2.2.1.1)
- 社内資料:健康成人における薬物動態(反復投与)(2010年4月16日承認、CTD2.7.2.2.2.1.1)
- 社内資料:生物学的同等性試験
- 社内資料:食事の影響(2010年4月16日承認、CTD2.7.1.2.2)
- 社内資料:放射性標識体投与時の薬物動態及び代謝(2010年4月16日承認、CTD2.7.2.2.1.6)
- 社内資料:血漿蛋白結合(2010年4月16日承認、CTD2.7.2.2.1.5)
- 社内資料:ヒトcytochrome P450に対する阻害作用(2010年4月16日承認、CTD2.7.2.2.1.3)
- 社内資料:高齢者における薬物動態(2010年4月16日承認、CTD2.7.2.2.2.2.1)
- 社内資料:腎機能障害患者及び血液透析患者における薬物動態(2010年4月16日承認、CTD2.7.2.2.2.2.2)
- 社内資料:健康被験者、帯状疱疹後神経痛患者、糖尿病性末梢神経障害に伴う疼痛を有する患者及び線維筋痛症患者における母集団薬物動態(2010年10月27日承認CTD2.7.2.3.5.1、2012年6月22日承認CTD2.7.2.3.1、2.7.2.3.4)
- 社内資料:ガバペンチンとの薬物相互作用(2010年4月16日承認、CTD2.7.2.2.2.3.5)
- 社内資料:経口避妊薬との薬物相互作用(2010年4月16日承認、CTD2.7.2.2.2.3.6)
- 社内資料:ロラゼパムとの薬物相互作用(2010年4月16日承認、CTD2.7.2.2.2.4.1)
- 社内資料:オキシコドンとの薬物相互作用(2010年4月16日承認、CTD2.7.2.2.2.4.2)
- 社内資料:エタノールとの薬物相互作用(2010年4月16日承認、CTD2.7.2.2.2.4.3)
- Brodie MJ,et al., Epilepsia., 46 (9), 1407-1413, (2005) »PubMed
- 小川節郎ほか, 日本ペインクリニック学会誌, 17 (2), 141-152, (2010) »DOI
- 社内資料:国内第III相検証試験(糖尿病性末梢神経障害に伴う疼痛)(2010年10月27日承認、CTD2.7.3.3)
- 社内資料:国内第III相検証試験(線維筋痛症)(2012年6月22日承認、CTD2.7.3.3)
- 社内資料:国内長期投与試験(帯状疱疹後神経痛)(2010年4月16日承認、CTD2.7.3.5.1)
- 社内資料:国内長期投与試験(糖尿病性末梢神経障害に伴う疼痛)(2010年10月27日承認、CTD2.7.3.5.2)
- 社内資料:国内長期投与試験(線維筋痛症)(2012年6月22日承認、CTD2.7.3.5.1)
- 社内資料:国内長期投与試験(脊髄損傷後疼痛、脳卒中後疼痛、多発性硬化症に伴う疼痛)(2013年2月28日承認、CTD2.7.3.5.1)
- 社内資料:国際共同第III相試験(脊髄損傷後疼痛)(2013年2月28日承認、CTD2.7.3.3)
- 社内資料:外国第II相及び第III相プラセボ対照試験(帯状疱疹後神経痛)(2010年4月16日承認、CTD2.7.3.3)
- 社内資料:外国長期投与試験(帯状疱疹後神経痛)(2010年4月16日承認、CTD2.7.3.5.2)
- Bauer CS,et al., J Neurosci., 29 (13), 4076-4088, (2009) »PubMed
- Fink K,et al., Neuropharmacology., 42 (2), 229-236, (2002) »PubMed
- Maneuf YP,et al., Pain., 93 (2), 191-196, (2001) »PubMed
- Tanabe M,et al., J Neurosci Res., 86 (15), 3258-3264, (2008) »PubMed
- Bee LA,et al., Pain., 140 (1), 209-223, (2008) »PubMed
- Field MJ,et al., Pain., 83 (2), 303-311, (1999) »PubMed
- Field MJ,et al., Pain., 80 (1-2), 391-398, (1999) »PubMed
- Tanabe M,et al., Eur J Pharmacol., 609 (1-3), 65-68, (2009) »PubMed
- 社内資料:慢性筋骨格系疼痛モデルにおける薬効薬理試験(2012年6月22日承認、CTD2.6.2.2)
- Field MJ,et al., Br J Pharmacol., 121 (8), 1513-1522, (1997) »PubMed
- Field MJ,et al., J Pharmacol Exp Ther., 282 (3), 1242-1246, (1997) »PubMed
24. 文献請求先及び問い合わせ先
文献請求先
ヴィアトリス製薬合同会社
メディカルインフォメーション部
〒106-0041
東京都港区麻布台一丁目3番1号
電話:フリーダイヤル 0120-419-043
製品情報問い合わせ先
ヴィアトリス製薬合同会社
メディカルインフォメーション部
〒106-0041
東京都港区麻布台一丁目3番1号
電話:フリーダイヤル 0120-419-043
26. 製造販売業者等
26.1 製造販売元
ヴィアトリス製薬合同会社
東京都港区麻布台一丁目3番1号