医薬品情報
| 総称名 | アビガン |
|---|---|
| 一般名 | ファビピラビル |
| 欧文一般名 | Favipiravir |
| 製剤名 | ファビピラビル錠 |
| 薬効分類名 | 抗ウイルス剤 |
| 薬効分類番号 | 6250 |
| ATCコード | J05AX27 |
| KEGG DRUG |
D09537
ファビピラビル
|
| KEGG DGROUP |
DG03029
抗インフルエンザウイルス薬
|
| JAPIC | 添付文書(PDF) |
添付文書情報2024年8月 改訂(第3版)

<新型又は再興型インフルエンザウイルス感染症>
本剤は、他の抗インフルエンザウイルス薬が無効又は効果不十分な新型又は再興型インフルエンザウイルス感染症が発生し、本剤を当該インフルエンザウイルスへの対策に使用すると国が判断した場合にのみ、患者への投与が検討される医薬品である。本剤の使用に際しては、国が示す当該インフルエンザウイルスへの対策の情報を含め、最新の情報を随時参照し、適切な患者に対して使用すること。
新型又は再興型インフルエンザウイルス感染症に対する本剤の投与経験はない。電子添文中の副作用、臨床成績等の情報については、承認用法及び用量より低用量で実施した国内臨床試験に加え海外での臨床成績に基づき記載している。
新型又は再興型インフルエンザウイルス感染症に対する本剤の投与経験はない。電子添文中の副作用、臨床成績等の情報については、承認用法及び用量より低用量で実施した国内臨床試験に加え海外での臨床成績に基づき記載している。
商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| アビガン錠200mg | AVIGAN Tablets 200mg | 富士フイルム富山化学 | 6250054F1022 | 39862.5円/錠 | 劇薬, 処方箋医薬品注) |
1. 警告
<重症熱性血小板減少症候群ウイルス感染症>
1.1 本剤は重症感染症診療体制が整備され、緊急時に十分な措置が可能な医療機関において、本剤について十分な知識をもつ医師のもと、入院管理下で投与すること。
<効能共通>
1.3 妊娠する可能性のある女性に投与する場合は、投与開始前に妊娠検査を行い、陰性であることを確認した上で、投与を開始すること。また、その危険性について十分に説明した上で、投与期間中及び投与終了後10日間はパートナーと共に極めて有効な避妊法の実施を徹底するよう指導すること。なお、本剤の投与期間中に妊娠が疑われる場合には、直ちに投与を中止し、医師等に連絡するよう患者を指導すること。[9.4.1参照]
<新型又は再興型インフルエンザウイルス感染症>
1.5 本剤の投与にあたっては、本剤の必要性を慎重に検討すること。
2. 禁忌
4. 効能または効果
○新型又は再興型インフルエンザウイルス感染症(ただし、他の抗インフルエンザウイルス薬が無効又は効果不十分なものに限る。)
5. 効能または効果に関連する注意
<新型又は再興型インフルエンザウイルス感染症>
5.1 本剤は、他の抗インフルエンザウイルス薬が無効又は効果不十分な新型又は再興型インフルエンザウイルス感染症が発生し、本剤を当該インフルエンザウイルスへの対策に使用すると国が判断した場合にのみ、患者への投与が検討される医薬品である。本剤の使用に際しては、国が示す当該インフルエンザウイルスへの対策の情報を含め、最新の情報を随時参照し、適切な患者に対して使用すること。
5.2 本剤は細菌感染症には効果がない。[8.3参照]
<効能共通>
5.3 小児等に対する投与経験はない。[9.7参照]
6. 用法及び用量
<新型又は再興型インフルエンザウイルス感染症>
通常、成人にはファビピラビルとして1日目は1回1600mgを1日2回、2日目から5日目は1回600mgを1日2回経口投与する。総投与期間は5日間とすること。
<重症熱性血小板減少症候群ウイルス感染症>
通常、成人にはファビピラビルとして1日目は1回1800mgを1日2回、2日目から10日目は1回800mgを1日2回経口投与する。総投与期間は10日間とすること。
7. 用法及び用量に関連する注意
<新型又は再興型インフルエンザウイルス感染症>
7.1 インフルエンザ様症状の発現後速やかに投与を開始すること。
<重症熱性血小板減少症候群ウイルス感染症>
7.3 重症熱性血小板減少症候群ウイルス感染症の症状の発現後速やかに投与を開始すること。
8. 重要な基本的注意
<効能共通>
<新型又は再興型インフルエンザウイルス感染症>
8.2 抗インフルエンザウイルス薬の服用の有無又は種類にかかわらず、インフルエンザ罹患時には、異常行動を発現した例が報告されている。
異常行動による転落等の万が一の事故を防止するための予防的な対応として、[1]異常行動の発現のおそれがあること、[2]自宅において療養を行う場合、少なくとも発熱から2日間、保護者等は転落等の事故に対する防止対策を講じること、について患者・家族に対し説明を行うこと。
なお、転落等の事故に至るおそれのある重度の異常行動については、就学以降の小児・未成年者の男性で報告が多いこと、発熱から2日間以内に発現することが多いこと、が知られている。[11.1.1参照]
異常行動による転落等の万が一の事故を防止するための予防的な対応として、[1]異常行動の発現のおそれがあること、[2]自宅において療養を行う場合、少なくとも発熱から2日間、保護者等は転落等の事故に対する防止対策を講じること、について患者・家族に対し説明を行うこと。
なお、転落等の事故に至るおそれのある重度の異常行動については、就学以降の小児・未成年者の男性で報告が多いこと、発熱から2日間以内に発現することが多いこと、が知られている。[11.1.1参照]
8.3 細菌感染症がインフルエンザウイルス感染症に合併したり、インフルエンザ様症状と混同されることがある。細菌感染症の場合及び細菌感染症が疑われる場合には、抗菌剤を投与するなど適切な処置を行うこと。[5.2参照]
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 痛風又は痛風の既往歴のある患者及び高尿酸血症のある患者
血中尿酸値が上昇し、痛風発作があらわれることがある。[11.2参照]
9.3 肝機能障害患者
9.3.1 重度の肝機能障害患者(Child-Pugh分類クラスC)
9.3.2 軽度及び中等度の肝機能障害患者(Child-Pugh分類クラスA及びB)
9.4 生殖能を有する者
9.4.1 妊娠する可能性のある女性
9.5 妊婦
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。本剤の主代謝物である水酸化体がヒト母乳中へ移行することが認められている。
9.7 小児等
9.8 高齢者
患者の状態を観察しながら投与すること。一般に生理機能が低下していることが多い。
10. 相互作用
相互作用序文
薬物代謝酵素用語
アルデヒドオキシダーゼ(AO)
薬物代謝酵素用語
キサンチンオキシダーゼ(XO)
薬物代謝酵素用語
CYP2C8
10.2 併用注意
| ピラジナミド | 血中尿酸値が上昇する。 ピラジナミド1.5g1日1回、本剤1200mg/400mg1日2回が投与されたとき、血中尿酸値は、ピラジナミド単独投与時及び本剤併用投与時でそれぞれ11.6mg/dL及び13.9mg/dLであった。 | 腎尿細管における尿酸の再吸収を相加的に促進させる。 |
| CYP2C8で代謝される薬剤 レパグリニド 等 [16.7.2参照] | 左記薬剤の血中濃度が上昇し、左記薬剤の副作用が発現するおそれがある。 | CYP2C8を阻害することにより、左記薬剤の血中濃度を上昇させる。 |
| テオフィリン6) [16.7.2参照] | 本剤の血中濃度が上昇し、本剤の副作用が発現するおそれがある。 | XOを介した相互作用により、本剤の血中濃度を上昇させることが考えられる。 |
| ファムシクロビル スリンダク | これらの薬剤の効果を減弱させるおそれがある。 | 本剤がAOを阻害する4)ことにより、これらの薬剤の活性化体の血中濃度を低下させることが考えられる。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 異常行動(頻度不明)
因果関係は不明であるものの、インフルエンザ罹患時には、転落等に至るおそれのある異常行動(急に走り出す、徘徊する等)があらわれることがある。[8.2参照]
11.1.2 ショック、アナフィラキシー(いずれも頻度不明)
11.1.3 肺炎(頻度不明)
11.1.4 劇症肝炎(頻度不明)、肝機能障害(0.2%)、黄疸(頻度不明)[8.1参照]
11.1.5 中毒性表皮壊死融解症(Toxic Epidermal Necrolysis:TEN)、皮膚粘膜眼症候群(Stevens-Johnson症候群)(いずれも頻度不明)
11.1.6 急性腎障害(頻度不明)
11.1.7 白血球減少、好中球減少、血小板減少(いずれも頻度不明)
11.1.8 痙攣(0.2%)、精神神経症状(意識障害、譫妄、幻覚、妄想等)(頻度不明)
11.1.9 出血性大腸炎(頻度不明)
11.2 その他の副作用
| 1%以上 | 0.5〜1%未満 | 0.5%未満 | 頻度不明 | |
| 過敏症 | 発疹 | − | 湿疹、そう痒症、紅斑 | − |
| 肝臓 | AST増加、ALT増加、γ-GTP増加 | − | 血中ALP増加、血中ビリルビン増加 | − |
| 腎臓 | − | 尿中ブドウ糖陽性 | 尿中血陽性 | − |
| 消化器 | 下痢(4.5%) | 悪心、腹痛、嘔吐 | 腹部不快感、胃炎、十二指腸潰瘍、血便排泄、口内炎 | − |
| 血液 | 好中球数減少、白血球数減少 | − | 白血球数増加、網状赤血球数減少、単球数増加、リンパ節症 | − |
| 代謝異常 | 血中尿酸増加(7.0%)注)、血中トリグリセリド増加 | − | 痛風注)、血中カリウム減少 | − |
| 呼吸器 | − | − | 喘息、口腔咽頭痛、鼻炎、鼻咽頭炎、誤嚥性肺炎 | − |
| その他 | − | − | 味覚異常、血中CK増加、心電図QT延長、扁桃腺ポリープ、蜂巣炎、霧視、眼痛、回転性めまい、上室性期外収縮、心室性期外収縮、心電図ST-T部分異常、心電図T波逆転、色素沈着、筋肉痛、挫傷 | 発熱 |
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
15. その他の注意
15.2 非臨床試験に基づく情報
16. 薬物動態
16.1 血中濃度
16.1.1 反復投与(1600mg/600mg BID)
健康成人8例に本剤を1日目は1回1600mgを1日2回、2日目から6日目は1回600mgを1日2回(6日目は1回のみ)経口投与(1600mg/600mg BID)したときの薬物動態パラメータは次のとおりであった。[7.2参照]
| 投与方法 | 例数 | Cmax注1)(μg/mL) | AUC注1)注2)(μg・hr/mL) | Tmax注3)(hr) | t1/2注4)(hr) | |
| 1600mg/600mg BID | 1日目 | 8 | 64.56 [17.2] | 446.09 [28.1] | 1.5 [0.75,4] | 4.8±1.1 |
| 6日目 | 8 | 64.69 [24.1] | 553.98 [31.2] | 1.5 [0.75,2] | 5.6±2.3 |
図1 本剤の血漿中濃度推移(平均値±標準偏差)
16.1.2 反復投与(1800mg/800mg BID)
健康成人8例に本剤を1日目は1回1800mgを1日2回、2日目から22日目は1回800mgを1日2回(22日目は1回のみ)経口投与(1800mg/800mg BID)したときの薬物動態パラメータは次のとおりであった9)。
| 投与方法 | 例数 | Cmax注5)(μg/mL) | AUC注5)注6)(μg・hr/mL) | Tmax注7)(hr) | t1/2注8)(hr) | |
| 1800mg/800mg BID | 1日目 | 8注9) | 65.06 [22.7] | 724.56 [47.1] | 1.5 [1,4] | 7.5±2.7 |
| 12日目 | 7 | 104.08 [21.3] | 966.41 [23.9] | 1.5 [0.5,2] | 17.6±7.4 | |
| 22日目 | 7 | 100.39 [21.3] | 932.44 [24.6] | 1.5 [0.75,2] | 8.1±2.6 |
図2 本剤の血漿中濃度推移(平均値±標準偏差)
16.1.3 アルデヒドオキシダーゼ活性が低いと考えられる者
アルデヒドオキシダーゼ(AO)活性がほとんどないと考えられる健康成人1例に本剤を7日間反復経口投与[本剤を1日目初回は1200mg、1日目2回目は400mg、2日目から6日目は1回400mgを1日2回、7日目は400mgを1回投与]注10)したとき、投与1日目及び投与7日目の未変化体のAUCの推定値は、それぞれ1452.73μg・hr/mL及び1324.09μg・hr/mLであった10)。
16.2 吸収
16.2.1 食事の影響
健康成人15例にクロスオーバー法により本剤1200mgを空腹時及び食後に単回経口投与注10)したところ、本剤の空腹時に対する食後投与のCmax及びAUCの幾何平均の比[90%信頼区間]は、それぞれ0.908[0.826,0.998]及び0.963[0.888,1.044]であり、比の90%信頼区間はあらかじめ定めた範囲内(0.80〜1.25)であった11)。
16.3 分布
16.3.1 精液への分布
健康成人男性20例に本剤を1日目は1回1200mgを1日2回、2日目から5日目は1回800mgを1日2回経口投与(1200mg/800mg BID)注10)したときの本剤の精液中濃度(幾何平均)は投与3日目及び投与終了後2日目でそれぞれ18.341μg/mL及び0.053μg/mLであり、投与終了後7日目にはすべての被験者で定量下限(0.02μg/mL)未満となった12)。また、精液/血漿中濃度比(平均値)は投与3日目及び投与終了後2日目でそれぞれ0.53及び0.45であった(外国人データ)。
16.3.2 血清蛋白結合率
本剤のヒト血清蛋白結合率は、0.3〜30μg/mLの濃度において、53.4〜54.4%であった(in vitro、遠心限外濾過法)。
16.3.3 動物でのデータ
サルに14C-ファビピラビルを単回経口投与したとき、各組織に広く移行した。各組織の放射能濃度は投与後0.5時間に最高値を示した後、血漿中放射能濃度と平行した推移を示した。投与後0.5時間の肺内放射能濃度の血漿中濃度比は0.51であり、投与後、呼吸器系組織に速やかに移行した。また、投与後0.5時間の腎臓中放射能濃度は血漿中よりも高く、血漿中濃度比は2.66であった。骨を除く各組織の放射能濃度は、投与後24時間までに最高濃度の2.8%以下に低下した13)。
16.4 代謝
16.5 排泄
本剤は主に水酸化体として尿中に排泄され、未変化体はわずかであった。健康成人6例に本剤を7日間反復経口投与[本剤を1日目初回は1200mg、1日目2回目は400mg、2日目から6日目は1回400mgを1日2回、7日目は400mgを1回投与]注10)したときの最終投与後48時間までの未変化体及び水酸化体の累積尿中排泄率は、それぞれ0.8%及び53.1%であった10)。
16.6 特定の背景を有する患者
16.6.1 肝機能障害患者
軽度及び中等度肝機能障害患者(Child-Pugh分類クラスA及びB、各6例)に、本剤を1日目は1回1200mgを1日2回、2日目から5日目は1回800mgを1日2回経口投与(1200mg/800mg BID)注10)したとき、投与5日目のCmax及びAUCは、健康成人に同様の用法及び用量で投与した場合と比べて、軽度肝機能障害患者ではそれぞれ約1.6倍及び約1.7倍、中等度肝機能障害患者ではそれぞれ約1.4倍及び約1.8倍であった。
重度肝機能障害患者(Child-Pugh分類クラスC、4例)に、本剤を1日目は1回800mgを1日2回、2日目から3日目は1回400mgを1日2回経口投与(800mg/400mg BID)注10)したとき、投与3日目のCmax及びAUCは、健康成人に同様の用法及び用量で投与した場合と比べて、それぞれ約2.1倍及び約6.3倍であった(外国人データ)14)。[9.3.1、9.3.2参照]
重度肝機能障害患者(Child-Pugh分類クラスC、4例)に、本剤を1日目は1回800mgを1日2回、2日目から3日目は1回400mgを1日2回経口投与(800mg/400mg BID)注10)したとき、投与3日目のCmax及びAUCは、健康成人に同様の用法及び用量で投与した場合と比べて、それぞれ約2.1倍及び約6.3倍であった(外国人データ)14)。[9.3.1、9.3.2参照]
16.6.2 腎機能障害患者
軽度、中等度及び重度腎機能障害患者(CLcr:60〜89mL/min、30〜59mL/min及び30mL/min未満の透析していない患者、各4例)に、本剤1800mgを単回経口投与注10)したとき、Cmax及びAUCinfは、本剤1800mgを単回経口投与した健康成人と比べて、軽度腎機能障害患者ではそれぞれ約1.0倍及び約1.2倍、中等度腎機能障害患者ではいずれも約0.9倍、重度腎機能障害患者ではそれぞれ約1.0倍及び約1.2倍であった。
本剤の代謝物である水酸化体のCmax及びAUCinfは、本剤1800mgを単回経口投与した健康成人と比べて、軽度腎機能障害患者ではそれぞれ約1.1倍及び約1.2倍、中等度腎機能障害患者ではそれぞれ約1.6倍及び約2.2倍、重度腎機能障害患者ではそれぞれ約2.5倍及び約6.5倍であった(外国人データ)15)。
本剤の代謝物である水酸化体のCmax及びAUCinfは、本剤1800mgを単回経口投与した健康成人と比べて、軽度腎機能障害患者ではそれぞれ約1.1倍及び約1.2倍、中等度腎機能障害患者ではそれぞれ約1.6倍及び約2.2倍、重度腎機能障害患者ではそれぞれ約2.5倍及び約6.5倍であった(外国人データ)15)。
注10)本剤の承認用法及び用量は、「1日目は1回1600mgを1日2回、2日目から5日目は1回600mgを1日2回経口投与」又は「1日目は1回1800mgを1日2回、2日目から10日目は1回800mgを1日2回経口投与」
16.7 薬物相互作用
16.7.1 非臨床薬物相互作用試験
16.7.2 臨床薬物相互作用試験
臨床薬物相互作用試験の結果は次のとおりであった。[10.2参照]
| 併用薬剤及び用量 | 本剤の用量 | 例数 | 投与時期 | 本剤の薬物動態パラメータの比 [90%信頼区間] (併用投与/単独投与) | |
| Cmax | AUC | ||||
| テオフィリン6) 1〜9日目に200mg1日2回、10日目に200mg1日1回 | 6日目に600mg1日2回、7〜10日目に600mg1日1回 | 10 | 6日目 | 1.33 [1.19,1.48] | 1.27 [1.15,1.40] |
| 7日目 | 1.03 [0.92,1.15] | 1.17 [1.04,1.31] | |||
| オセルタミビル16) 1〜5日目に75mg1日2回、6日目に75mg1日1回 | 5日目に600mg1日2回、6日目に600mg1日1回 | 10 | 6日目 | 0.98 [0.87,1.10] | 1.01 [0.91,1.11] |
| ラロキシフェン 1〜3日目に60mg1日1回注11) | 1日目に1200mg1日2回、2日目に800mg1日2回、3日目に800mg1日1回 | 17 | 1日目 | 1.00 [0.90,1.10] | 1.03 [0.95,1.12] |
| 3日目 | 0.90 [0.81,0.99] | 0.85 [0.79,0.93] | |||
| ヒドララジン 1、5日目に5mg1日1回 | 1日目初回に1200mg、2回目に400mg、2〜4日目に400mg1日2回、5日目に400mg1日1回 | 14 | 1日目 | 0.99 [0.92,1.06] | 0.99 [0.92,1.07] |
| 5日目 | 0.96 [0.89,1.04] | 1.04 [0.96,1.12] | |||
| 併用薬剤及び用量 | 本剤の用量 | 例数 | 投与時期 | 併用薬剤の薬物動態パラメータの比 [90%信頼区間] (併用投与/単独投与) | |
| Cmax | AUC | ||||
| テオフィリン6) 1〜9日目に200mg1日2回、10日目に200mg1日1回 | 6日目に600mg1日2回、7〜10日目に600mg1日1回 | 10 | 7日目 | 0.93 [0.85,1.01] | 0.92 [0.87,0.97] |
| 10日目 | 0.99 [0.94,1.04] | 0.97 [0.91,1.03] | |||
| オセルタミビル16) 1〜5日目に75mg1日2回、6日目に75mg1日1回 | 5日目に600mg1日2回、6日目に600mg1日1回 | 10 | 6日目 | 1.10 [1.06,1.15] | 1.14 [1.10,1.18] |
| アセトアミノフェン 1、5日目に650mg1日1回注11) | 1日目に1200mg1日2回、2〜4日目に800mg1日2回、5日目に800mg1日1回 | 28 | 1日目 | 1.03 [0.93,1.14] | 1.16 [1.08,1.25] |
| 5日目 | 1.08 [0.96,1.22] | 1.14 [1.04,1.26] | |||
| ノルエチンドロン/エチニルエストラジオール配合剤 1〜5日目に1mg/0.035mg1日1回注11) | 1日目に1200mg1日2回、2〜4日目に800mg1日2回、5日目に800mg1日1回 | 25 | 12日目注12) | 1.23 [1.16,1.30] | 1.47 [1.42,1.52] |
| 12日目注13) | 1.48 [1.42,1.54] | 1.43 [1.39,1.47] | |||
| レパグリニド 13日目に0.5mg1日1回注11) | 1日目に1200mg1日2回、2〜4日目に800mg1日2回、5日目に800mg1日1回 | 17 | 13日目 | 1.28 [1.16,1.41] | 1.52 [1.37,1.68] |
| ヒドララジン 1、5日目に5mg1日1回 | 1日目初回に1200mg、2回目に400mg、2〜4日目に400mg1日2回、5日目に400mg1日1回 | 14 | 1日目 | 0.73 [0.67,0.81] | 0.87 [0.78,0.97] |
| 5日目 | 0.79 [0.71,0.88] | 0.91 [0.82,1.01] | |||
| トリアゾラム 1、4日目に0.25mg1日1回 | 3日目に1800mg1日2回、4日目に800mg1日2回 | 12 | 4日目 | 1.12 [0.89,1.42] | 1.01 [0.91,1.11] |
| メトホルミン 1、4日目に250mg1日1回 | 3日目に1800mg1日2回、4日目に800mg1日2回 | 12 | 4日目 | 0.96 [0.87,1.07] | 1.01 [0.94,1.09] |
17. 臨床成績
17.1 有効性及び安全性に関する試験
<新型又は再興型インフルエンザウイルス感染症>
17.1.1 海外第I/II相試験
A型又はB型インフルエンザウイルス感染症患者(成人)を対象として、プラセボを対照とした第I/II相試験[本剤を1日目は1回1800mgを1日2回、2日目から5日目は1回800mgを1日2回経口投与(1800mg/800mg BID)及び本剤を1日目初回は2400mg、2回目及び3回目は1回600mg、2日目から5日目は1回600mgを1日3回経口投与(2400mg/600mg TID)]注1)を実施した。主要評価項目である罹病期間注2)について、プラセボ群(88例)と本剤1800mg/800mg BID群(101例)との対比較では、統計学的に有意な差が認められたが(p=0.01、Gehan-Wilcoxon test)、本剤2400mg/600mg TID群(82例)との対比較では、統計学的に有意な差は認められなかった(p=0.414、Gehan-Wilcoxon test)。本剤群において、副作用は認められなかった。[7.2参照]
図1 インフルエンザ主要症状罹病期間
17.1.2 海外第III相試験
A型又はB型インフルエンザウイルス感染症患者(成人)を対象として、プラセボを対照とした第III相試験[本剤を1日目は1800mgを1日2回、2日目から5日目は1回800mgを1日2回経口投与(1800mg/800mg BID)]注1)について、主要評価項目をインフルエンザ主要症状罹病期間注3)と設定し、実施した結果は以下のとおりであった。
| 本剤群(301例) | プラセボ群(322例) | |
| イベント数 | 288 | 306 |
| 中央値[95%信頼区間](時間) | 84.2[77.1,95.7] | 98.6[94.6,107.1] |
| p値 (Peto-Peto-Prentice検定) | 0.004 | |
図2 主要評価項目注3)に係るKaplan-Meierプロット図(ITTI集団)
副作用発現頻度は、本剤群で7.9%(34/428例)で、主な副作用は、浮動性めまい1.2%(5/428例)であった。
17.1.3 海外第III相試験
A型又はB型インフルエンザウイルス感染症患者(成人)を対象として、プラセボを対照とした第III相試験[本剤を1日目は1800mgを1日2回、2日目から5日目は1回800mgを1日2回経口投与(1800mg/800mg BID)]注1)について、主要評価項目をインフルエンザ主要症状罹病期間注3)と設定し、実施した結果は以下のとおりであった。
| 本剤群(526例) | プラセボ群(169例) | |
| イベント数 | 505 | 163 |
| 中央値[95%信頼区間](時間) | 77.8[72.3,82.5] | 83.9[76.0,95.5] |
| p値 (Peto-Peto-Prentice検定) | 0.303 | |
図3 主要評価項目注3)に係るKaplan-Meierプロット図(ITTI集団)
副作用発現頻度は、本剤群で10.2%(88/861例)で、主な副作用は、血中トリグリセリド増加2.0%(17/861例)、悪心1.5%(13/861例)、下痢1.3%(11/861例)であった。
17.1.4 国際共同第III相試験(参考)
A型又はB型インフルエンザウイルス感染症患者(成人)を対象として、オセルタミビルリン酸塩(1回75mg1日2回、5日間)を対照とした国際共同第III相試験[本剤を1日目初回は1200mg、1日目2回目は400mg、2日目から5日目は1回400mgを1日2回経口投与]注1)を実施した[757例(日本540例、韓国78例、台湾139例)]。インフルエンザ主要症状罹病期間注4)の中央値[95%信頼区間]は、本剤群(377例)で63.1[55.5,70.4]時間、オセルタミビルリン酸塩群(380例)で51.2[45.9,57.6]時間であり、オセルタミビルリン酸塩群に対する本剤群のハザード比[95%信頼区間]は、0.818[0.707,0.948]であり、本剤の有効性は示されなかった(p=0.007、log-rank test)。
副作用発現頻度は、本剤群で19.8%(75/378例)であった。主な副作用は、血中尿酸増加5.6%(21/378例)、下痢4.2%(16/378例)、血中トリグリセリド増加1.9%(7/378例)であった。
17.1.5 海外第II相試験(参考)
A型又はB型インフルエンザウイルス感染症患者(成人)を対象として、プラセボを対照とした海外第II相試験[本剤を1日目は1回1000mgを1日2回、2日目から5日目は1回400mgを1日2回経口投与(1000mg/400mg BID)、本剤を1日目は1回1200mgを1日2回、2日目から5日目は1回800mgを1日2回経口投与(1200mg/800mg BID)及びプラセボを1日2回経口投与]注1)を実施した。インフルエンザ主要症状罹病期間注5)の中央値[95%信頼区間]は、本剤1000mg/400mg BID群(88例)で100.4[82.4,119.8]時間、本剤1200mg/800mg BID群(121例)で86.5[79.2,102.1]時間、プラセボ群(124例)で91.9[70.3,105.4]時間であり、プラセボ群との対比較において、本剤群のいずれにおいても、統計学的に有意な差は認められなかった(p>0.05、Gehan-Wilcoxon test、検定の多重性はStep-down法で調整)。
副作用発現頻度は、本剤1000mg/400mg BID群で18.9%(25/132例)、本剤1200mg/800mg BID群で19.6%(37/189例)であった。主な副作用は、本剤1000mg/400mg BID群で下痢2.3%(3/132例)、血中尿酸増加2.3%(3/132例)、本剤1200mg/800mg BID群で下痢3.2%(6/189例)、血中尿酸増加3.2%(6/189例)であった。
注1)本剤の新型又は再興型インフルエンザウイルス感染症に対する承認用法及び用量は、「1日目は1回1600mgを1日2回、2日目から5日目は1回600mgを1日2回経口投与」
注2)インフルエンザ主要6症状(咳嗽、咽頭痛、頭痛、鼻閉、筋肉痛、全身倦怠感)及び発熱の持続時間
注3)インフルエンザ主要6症状(咳嗽、咽頭痛、頭痛、鼻閉、筋肉痛、全身倦怠感)及び発熱が「改善」するまでの時間。インフルエンザ主要6症状のすべてが消失あるいは軽度となり、かつ発熱が回復した状態を21.5時間持続した場合を「改善」と定義。
注4)治験薬投与開始後から7つのインフルエンザ主要症状[咳嗽、咽喉頭痛、頭痛、鼻閉、熱感、筋肉痛及び全身倦怠感]がすべて「改善」するまでの時間(すべてのスコアが「1」以下に達した時点)。患者日誌をもとに治験責任医師又は治験分担医師がスコア化したインフルエンザ症状が「1」以下となってから21.5時間以上そのスコアを維持した状態を「改善」と定義。
注5)インフルエンザ主要6症状(咳嗽、咽頭痛、頭痛、鼻閉、筋肉痛、全身倦怠感)がすべて「改善」するまでの時間(すべてのスコアが「1」以下に低下した時点)及び発熱が20歳以上65歳未満の患者では38℃以下、65歳以上の患者では37.8℃以下を21.5時間以上維持した状態。
<重症熱性血小板減少症候群ウイルス感染症>
17.1.6 国内第III相試験
重症熱性血小板減少症候群(SFTS)患者を対象として、国内第III相試験[本剤を1日目は1回1800mgを1日2回、2日目から10日目は1回800mgを1日2回経口投与]を実施した。主たる有効性解析対象例(mITTE)注6)における、主要評価項目である本剤投与開始から28日目までの累積致死率は15.8%(3/19例)[95%信頼区間:3.4,39.6%]であり、事前設定された致死率の閾値(12.5%)注7)の点推定値を上回った。
副作用発現頻度は70.0%(21/30例)で、主な副作用は、高尿酸血症23.3%(7/30例)、血中尿酸増加20.0%(6/30例)、高トリグリセリド血症10.0%(3/30例)、発疹6.7%(2/30例)、心電図QT延長6.7%(2/30例)であった。
注6)国立感染症研究所で実施したRT-PCR検査でSFTSウイルスが検出され、症状発症後5日以内(144時間以内)に本剤を投与開始した症例(modified intention-to-treat evaluable)
注7)2015年1月から2017年9月における国立感染症研究所の感染症発生動向調査の累積致死率(2016年に実施した医師主導臨床研究で本剤を投与した10例を除く)
18. 薬効薬理
18.1 作用機序
18.2 In vitro抗ウイルス活性
A型及びB型インフルエンザウイルス実験室株に対するEC50値は、0.014〜0.55μg/mLであり、抗ウイルス活性を示した。
アダマンタン(アマンタジン及びリマンタジン)、オセルタミビル及びザナミビル耐性株を含む季節性のA型及びB型インフルエンザウイルスに対するEC50値は、それぞれ0.03〜0.94μg/mL及び0.09〜0.83μg/mLであった。
豚由来A型及び高病原性株を含む鳥由来A型(H5N1、H7N9株を含む)をはじめとするA型インフルエンザウイルス(アダマンタン、オセルタミビル及びザナミビル耐性株を含む)に対するEC50値は、0.06〜3.53μg/mLであった。
アダマンタン、オセルタミビル及びザナミビル全てに耐性のA型及びB型インフルエンザウイルスに対するEC50値は0.09〜0.47μg/mLであり、交差耐性を示さなかった19)20)。
SFTSウイルスの各種臨床分離株(J1型、J2型、J3型、C3型、C4型及びC5型)に対して抗ウイルス活性を示し、EC90値は14.83〜38.73μmol/L(2.33〜6.08μg/mL)、EC99値は48.20〜79.40μmol/L(7.57〜12.47μg/mL)であった。
アダマンタン(アマンタジン及びリマンタジン)、オセルタミビル及びザナミビル耐性株を含む季節性のA型及びB型インフルエンザウイルスに対するEC50値は、それぞれ0.03〜0.94μg/mL及び0.09〜0.83μg/mLであった。
豚由来A型及び高病原性株を含む鳥由来A型(H5N1、H7N9株を含む)をはじめとするA型インフルエンザウイルス(アダマンタン、オセルタミビル及びザナミビル耐性株を含む)に対するEC50値は、0.06〜3.53μg/mLであった。
アダマンタン、オセルタミビル及びザナミビル全てに耐性のA型及びB型インフルエンザウイルスに対するEC50値は0.09〜0.47μg/mLであり、交差耐性を示さなかった19)20)。
SFTSウイルスの各種臨床分離株(J1型、J2型、J3型、C3型、C4型及びC5型)に対して抗ウイルス活性を示し、EC90値は14.83〜38.73μmol/L(2.33〜6.08μg/mL)、EC99値は48.20〜79.40μmol/L(7.57〜12.47μg/mL)であった。
18.3 動物モデルにおける治療効果
インフルエンザウイルスA(H7N9)、A(H1N1)pdm09及びA(H3N2)によるマウス感染モデルにおいて、60mg/kg/日以下の5日間経口投与により肺内ウイルス量を低下させた21)22)23)。
インフルエンザウイルスA(H3N2)及びA(H5N1)によるマウス感染モデルにおいて、30mg/kg/日の5日間経口投与により治療効果を示した19)23)。
また、インフルエンザウイルスA(H3N2)による重症複合型免疫不全マウス感染モデルにおいて、30mg/kg/日の14日間の経口投与により治療効果を示した24)。
SFTSウイルスによるマウス感染モデルにおいて、120mg/kg/日及び200mg/kg/日の5日間経口投与により、生存率及び体重変化を指標とする治療効果を示し、血中ウイルスRNA量を低下させた25)。
インフルエンザウイルスA(H3N2)及びA(H5N1)によるマウス感染モデルにおいて、30mg/kg/日の5日間経口投与により治療効果を示した19)23)。
また、インフルエンザウイルスA(H3N2)による重症複合型免疫不全マウス感染モデルにおいて、30mg/kg/日の14日間の経口投与により治療効果を示した24)。
SFTSウイルスによるマウス感染モデルにおいて、120mg/kg/日及び200mg/kg/日の5日間経口投与により、生存率及び体重変化を指標とする治療効果を示し、血中ウイルスRNA量を低下させた25)。
18.4 耐性
ファビピラビル存在下で30代まで継代したA型インフルエンザウイルスのファビピラビルに対する感受性に変化はなく、耐性ウイルスは選択されなかった19)。なお、国際共同第III相試験をはじめとする臨床試験において、本剤耐性インフルエンザウイルスの出現状況に関する情報は得られていない。
ファビピラビル存在下で10代まで継代したSFTSウイルスにおいて、ファビピラビルに対する感受性の低下は観察されず、耐性ウイルスは選択されなかった。なお、国内第III相試験において、本剤耐性SFTSウイルスの出現状況に関する情報は得られていない。
ファビピラビル存在下で10代まで継代したSFTSウイルスにおいて、ファビピラビルに対する感受性の低下は観察されず、耐性ウイルスは選択されなかった。なお、国内第III相試験において、本剤耐性SFTSウイルスの出現状況に関する情報は得られていない。
19. 有効成分に関する理化学的知見
19.1. ファビピラビル
| 一般的名称 | ファビピラビル |
|---|---|
| 一般的名称(欧名) | Favipiravir |
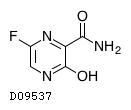
| 化学名 | 6-Fluoro-3-hydroxypyrazine-2-carboxamide |
| 分子式 | C5H4FN3O2 |
| 分子量 | 157.10 |
| 融点 | 187〜193℃ |
| 物理化学的性状 | 白色〜淡黄色の粉末である。アセトニトリル又はメタノールにやや溶けにくく、水又はエタノール(99.5)に溶けにくい。 |
| KEGG DRUG |
|
21. 承認条件
<効能共通>
21.1 医薬品リスク管理計画を策定の上、適切に実施すること。
21.2 本剤が承認されている効能又は効果(他の抗インフルエンザウイルス薬が無効又は効果不十分な新型又は再興型インフルエンザウイルス感染症にあっては、当該感染症への対策に使用すると厚生労働大臣が判断した場合に限る。)においてのみ使用されるよう、厳格な流通管理及び十分な安全対策を実施すること。
<新型又は再興型インフルエンザウイルス感染症>
21.3 本剤の使用実態下における有効性及び安全性について十分な検討が必要であることから、適切な製造販売後調査等を実施すること。
<重症熱性血小板減少症候群ウイルス感染症>
21.4 国内での治験症例が極めて限られていることから、製造販売後、一定数の症例に係るデータが集積されるまでの間は、全症例を対象に使用成績調査を実施することにより、本剤の使用患者の背景情報を把握するとともに、本剤の安全性及び有効性に関するデータを早期に収集し、本剤の適正使用に必要な措置を講じること。
21.5 本剤の重症熱性血小板減少症候群ウイルス感染症に対する使用に関する十分な知識・経験を持つ医師によってのみ処方・使用されるよう、製造販売にあたって必要な措置を講じること。
22. 包装
90錠[10錠(PTP)×9]
100錠[10錠(PTP)×10]
23. 主要文献
- 社内資料:生殖発生毒性試験・ラット(承認年月日:2014年3月24日、CTD2.6.6.6.1、2.6.6.6.2)
- 社内資料:生殖発生毒性試験・マウスほか(承認年月日:2014年3月24日、CTD2.6.6.6)
- 社内資料:毒性試験・幼若イヌほか(承認年月日:2014年3月24日、CTD2.6.6.9.4.3)
- 社内資料:代謝(承認年月日:2014年3月24日、CTD2.6.4.5.3、2.6.4.7)
- 社内資料:薬物相互作用(承認年月日:2014年3月24日、CTD2.6.4.7、2.6.4.8)
- 社内資料:テオフィリン併用試験(承認年月日:2014年3月24日、CTD2.7.6.6.1)
- 社内資料:毒性試験・イヌ(承認年月日:2014年3月24日、CTD2.6.6.3.3)
- 社内資料:精巣毒性試験・マウスほか(承認年月日:2014年3月24日、CTD2.6.6.9.4.2)
- 社内資料:22日間反復投与試験
- 社内資料:高用量反復投与試験(承認年月日:2014年3月24日、CTD2.7.6.4.5)
- 社内資料:食事の影響試験(承認年月日:2014年3月24日、CTD2.7.6.1.2)
- 社内資料:精液移行性試験(承認年月日:2014年3月24日、CTD2.7.6.7.3)
- 社内資料:体内動態・動物(承認年月日:2014年3月24日、CTD2.6.4.4)
- 社内資料:肝機能障害患者の薬物動態
- 社内資料:腎機能障害患者の薬物動態
- 社内資料:オセルタミビル併用試験(承認年月日:2014年3月24日、CTD2.7.6.6.2)
- Furuta Y,et al., Antimicrob.Agents Chemother., 49, 981-986, (2005) »PubMed
- Yamada H,et al., Viruses., 13 (6), 1061-1073, (2021) »PubMed
- 高橋和美ほか, 医学と薬学, 66, 429-441, (2011)
- 社内資料:抗ウイルス活性と交差耐性(承認年月日:2014年3月24日、CTD2.6.2.2.1)
- Ito Y,et al., Nature., 460, 1021-1025, (2009)
- Watanabe T,et al., Nature., 501, 551-555, (2013) »PubMed
- 社内資料:治療効果・マウス(承認年月日:2014年3月24日、CTD2.6.2.2.2)
- 社内資料:治療効果・免疫不全マウス(承認年月日:2014年3月24日、CTD2.6.2.2.2.6)
- Tani H,et al., PLoS One., 13 (10), e0206416, (2018) »PubMed
24. 文献請求先及び問い合わせ先
文献請求先
富士フイルム富山化学株式会社
製品情報センター
〒104-0031
東京都中央区京橋2-14-1 兼松ビル
電話:0120-502-620
製品情報問い合わせ先
富士フイルム富山化学株式会社
製品情報センター
〒104-0031
東京都中央区京橋2-14-1 兼松ビル
電話:0120-502-620
25. 保険給付上の注意
本剤は「重症熱性血小板減少症候群ウイルス感染症」に対して使用した場合にのみ保険給付されます。
26. 製造販売業者等
26.1 製造販売元
富士フイルム富山化学株式会社
〒104-0031
東京都中央区京橋2-14-1 兼松ビル