医薬品情報
| 総称名 | リムパーザ |
|---|---|
| 一般名 | オラパリブ |
| 欧文一般名 | Olaparib |
| 製剤名 | オラパリブ錠 |
| 薬効分類名 | 抗悪性腫瘍剤 ポリアデノシン5'二リン酸リボースポリメラーゼ(PARP)阻害剤 |
| 薬効分類番号 | 4291 |
| ATCコード | L01XK01 |
| KEGG DRUG |
D09730
オラパリブ
|
| KEGG DGROUP |
DG02942
PARP阻害薬
|
| JAPIC | 添付文書(PDF) |
添付文書情報2026年3月 改訂(第7版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| リムパーザ錠100mg | Lynparza Tablets 100mg | アストラゼネカ | 4291052F1027 | 3225.1円/錠 | 劇薬, 処方箋医薬品注) |
| リムパーザ錠150mg | Lynparza Tablets 150mg | アストラゼネカ | 4291052F2023 | 4788円/錠 | 劇薬, 処方箋医薬品注) |
1. 警告
本剤は、緊急時に十分対応できる医療施設において、がん化学療法に十分な知識・経験を持つ医師のもとで、本剤の使用が適切と判断される症例についてのみ投与すること。また、治療開始に先立ち、患者又はその家族に有効性及び危険性を十分説明し、同意を得てから投与すること。
2. 禁忌
次の患者には投与しないこと
本剤の成分に対し過敏症の既往歴のある患者
4. 効能または効果
○白金系抗悪性腫瘍剤感受性の再発卵巣癌における維持療法
○BRCA遺伝子変異陽性の卵巣癌における初回化学療法後の維持療法
○相同組換え修復欠損を有する卵巣癌におけるベバシズマブ(遺伝子組換え)を含む初回化学療法後の維持療法
○がん化学療法歴のあるBRCA遺伝子変異陽性かつHER2陰性の手術不能又は再発乳癌
○BRCA遺伝子変異陽性かつHER2陰性で再発高リスクの乳癌における術後薬物療法
○BRCA遺伝子変異陽性の遠隔転移を有する去勢抵抗性前立腺癌
○BRCA遺伝子変異陽性の治癒切除不能な膵癌における白金系抗悪性腫瘍剤を含む化学療法後の維持療法
○ミスマッチ修復機能正常(pMMR)の進行・再発の子宮体癌におけるデュルバルマブ(遺伝子組換え)を含む化学療法後の維持療法
5. 効能または効果に関連する注意
<白金系抗悪性腫瘍剤感受性の再発卵巣癌における維持療法>
5.1 再発時の白金系抗悪性腫瘍剤を含む化学療法で奏効が維持されている患者を対象とすること。
<BRCA遺伝子変異陽性の卵巣癌における初回化学療法後の維持療法>
5.3 国際産婦人科連合(FIGO)進行期分類III期又はIV期の卵巣癌と診断され、白金系抗悪性腫瘍剤を含む初回化学療法で奏効が維持されている患者を対象とすること。
5.4 承認された体外診断用医薬品又は医療機器注)を用いた検査により、BRCA遺伝子変異を有することが確認された患者に投与すること。
5.5 臨床試験に組み入れられた患者における前治療歴等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、適応患者の選択を行うこと。[17.1.1参照]
<相同組換え修復欠損を有する卵巣癌におけるベバシズマブ(遺伝子組換え)を含む初回化学療法後の維持療法>
5.6 国際産婦人科連合(FIGO)進行期分類III期又はIV期の卵巣癌と診断され、白金系抗悪性腫瘍剤及びベバシズマブ(遺伝子組換え)を含む初回化学療法で奏効が維持されている患者を対象とすること。
5.7 承認された体外診断用医薬品又は医療機器注)を用いた検査により、相同組換え修復欠損を有することが確認された患者に投与すること。
<がん化学療法歴のあるBRCA遺伝子変異陽性かつHER2陰性の手術不能又は再発乳癌>
5.8 本剤の投与を行う場合には、アントラサイクリン系抗悪性腫瘍剤及びタキサン系抗悪性腫瘍剤を含む化学療法歴のある患者を対象とすること。
5.9 承認された体外診断用医薬品又は医療機器注)を用いた検査により、BRCA遺伝子変異を有することが確認された患者に投与すること。
<BRCA遺伝子変異陽性かつHER2陰性で再発高リスクの乳癌における術後薬物療法>
5.10 本剤の術前薬物療法としての有効性及び安全性は確立していない。
5.11 臨床試験に組み入れられた患者の再発高リスクの定義、前治療歴等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、適応患者の選択を行うこと。[17.1.6参照]
5.12 承認された体外診断用医薬品又は医療機器注)を用いた検査により、BRCA遺伝子変異を有することが確認された患者に投与すること。
<BRCA遺伝子変異陽性の遠隔転移を有する去勢抵抗性前立腺癌>
5.13 本剤の術後補助療法としての有効性及び安全性は確立していない。
5.14 承認された体外診断用医薬品又は医療機器注)を用いた検査により、BRCA遺伝子変異を有することが確認された患者に投与すること。
<BRCA遺伝子変異陽性の治癒切除不能な膵癌における白金系抗悪性腫瘍剤を含む化学療法後の維持療法>
5.16 本剤の手術の補助療法としての有効性及び安全性は確立していない。
5.17 白金系抗悪性腫瘍剤を含む化学療法で疾患進行が認められていない患者を対象とすること。
5.18 臨床試験に組み入れられた患者の病期、白金系抗悪性腫瘍剤を含む化学療法の投与期間等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、適応患者の選択を行うこと。[17.1.9参照]
5.19 承認された体外診断用医薬品又は医療機器注)を用いた検査により、生殖細胞系列のBRCA遺伝子変異(病的変異又は病的変異疑い)を有することが確認された患者に投与すること。
<ミスマッチ修復機能正常(pMMR)の進行・再発の子宮体癌におけるデュルバルマブ(遺伝子組換え)を含む化学療法後の維持療法>
5.20 デュルバルマブ(遺伝子組換え)及び白金系抗悪性腫瘍剤を含む化学療法で疾患進行が認められていない患者を対象とすること。
5.21 十分な経験を有する病理医又は検査施設における検査により、pMMRが確認された患者に投与すること。検査にあたっては、承認された体外診断用医薬品又は医療機器注)を用いること。
注)承認された体外診断用医薬品又は医療機器に関する情報については、以下のウェブサイトから入手可能である:
https://www.pmda.go.jp/review-services/drug-reviews/review-information/cd/0001.html
6. 用法及び用量
<白金系抗悪性腫瘍剤感受性の再発卵巣癌における維持療法、BRCA遺伝子変異陽性の卵巣癌における初回化学療法後の維持療法、BRCA遺伝子変異陽性の治癒切除不能な膵癌における白金系抗悪性腫瘍剤を含む化学療法後の維持療法>
通常、成人にはオラパリブとして1回300mgを1日2回、経口投与する。なお、患者の状態により適宜減量する。
<相同組換え修復欠損を有する卵巣癌におけるベバシズマブ(遺伝子組換え)を含む初回化学療法後の維持療法>
ベバシズマブ(遺伝子組換え)との併用において、通常、成人にはオラパリブとして1回300mgを1日2回、経口投与する。なお、患者の状態により適宜減量する。
<がん化学療法歴のあるBRCA遺伝子変異陽性かつHER2陰性の手術不能又は再発乳癌、BRCA遺伝子変異陽性かつHER2陰性で再発高リスクの乳癌における術後薬物療法>
通常、成人にはオラパリブとして1回300mgを1日2回、経口投与する。ただし、術後薬物療法の場合、投与期間は1年間までとする。なお、患者の状態により適宜減量する。
<BRCA遺伝子変異陽性の遠隔転移を有する去勢抵抗性前立腺癌>
通常、成人にはオラパリブとして1回300mgを1日2回、経口投与する。他の薬剤と併用する場合は、アビラテロン酢酸エステル及びプレドニゾロンと併用すること。なお、患者の状態により適宜減量する。
<ミスマッチ修復機能正常(pMMR)の進行・再発の子宮体癌におけるデュルバルマブ(遺伝子組換え)を含む化学療法後の維持療法>
デュルバルマブ(遺伝子組換え)との併用において、通常、成人にはオラパリブとして1回300mgを1日2回、経口投与する。なお、患者の状態により適宜減量する。
7. 用法及び用量に関連する注意
<効能共通>
7.1 100mg錠と150mg錠の生物学的同等性は示されていないため、300mgを投与する際には100mg錠を使用しないこと。
7.2 本剤投与により副作用が発現した場合には、以下の基準を考慮して、休薬・減量・中止すること。
| 副作用 | 程度注 | 処置 | 再開時の投与量 |
| 貧血 | ヘモグロビン値がGrade 3又は4の場合 | ヘモグロビン値≧9g/dLに回復するまで最大4週間休薬する。 | ・1回目の再開の場合、減量せずに投与する。 ・2回目の再開の場合、1回250mgを1日2回で投与する。 ・3回目の再開の場合、1回200mgを1日2回で投与する。 |
| 好中球減少 | Grade 3又は4の場合 | Grade 1以下に回復するまで休薬する。 | |
| 血小板減少 | Grade 3又は4の場合 | Grade 1以下に回復するまで最大4週間休薬する。 | 減量せずに投与する。 |
| 間質性肺疾患 | Grade 2の場合 | Grade 1以下に回復するまで休薬する。 | 減量せずに投与する。 |
| Grade 3又は4の場合 | 中止する。 | 再開しない。 | |
| デュルバルマブ(遺伝子組換え)との併用投与下の赤芽球癆 | 全Grade | 本剤及びデュルバルマブ(遺伝子組換え)の投与を中止する。 | 再開しない。 |
| デュルバルマブ(遺伝子組換え)との併用投与下の自己免疫性溶血性貧血 | 全Grade | 本剤及びデュルバルマブ(遺伝子組換え)の投与を中止する。 | 再開しない。 |
| 上記以外の副作用 | Grade 3又は4の場合 | Grade 1以下に回復するまで休薬する。 | 減量せずに投与する。 |
<白金系抗悪性腫瘍剤感受性の再発卵巣癌における維持療法、がん化学療法歴のあるBRCA遺伝子変異陽性かつHER2陰性の手術不能又は再発乳癌、BRCA遺伝子変異陽性の治癒切除不能な膵癌における白金系抗悪性腫瘍剤を含む化学療法後の維持療法>
7.3 他の抗悪性腫瘍剤との併用について、有効性及び安全性は確立していない。
<BRCA遺伝子変異陽性の卵巣癌における初回化学療法後の維持療法>
7.4 他の抗悪性腫瘍剤との併用について、有効性及び安全性は確立していない。
7.5 本剤の投与開始後2年が経過した時点で完全奏効が得られている患者においては、本剤の投与を中止すること。
<相同組換え修復欠損を有する卵巣癌におけるベバシズマブ(遺伝子組換え)を含む初回化学療法後の維持療法>
7.6 本剤の投与開始後2年が経過した時点で完全奏効が得られている患者においては、本剤の投与を中止すること。
7.7 ベバシズマブ(遺伝子組換え)の投与期間等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で投与すること。[17.1.4参照]
<BRCA遺伝子変異陽性かつHER2陰性で再発高リスクの乳癌における術後薬物療法>
7.8 他の抗悪性腫瘍剤との併用について、有効性及び安全性は確立していない。
7.9 内分泌療法剤との併用の必要性について、「17.臨床成績」の項の内容を熟知し、ホルモン受容体の発現状態等を考慮した上で判断すること。[17.1.6参照]
<BRCA遺伝子変異陽性の遠隔転移を有する去勢抵抗性前立腺癌>
7.10 アビラテロン酢酸エステル又はエンザルタミドによる治療歴のない患者における本剤単独投与の有効性及び安全性は確立していない。
7.11 他の抗悪性腫瘍剤との併用について、有効性及び安全性は確立していない。
7.12 外科的又は内科的去勢術と併用しない場合の有効性及び安全性は確立していない。
8. 重要な基本的注意
8.1 骨髄抑制があらわれることがあるので、本剤投与開始前及び投与中は定期的に血液検査を行い、患者の状態を十分に観察すること。[11.1.1参照]
9. 特定の背景を有する患者に関する注意
9.2 腎機能障害患者
減量を考慮するとともに、患者の状態をより慎重に観察し、有害事象の発現に十分注意すること。本剤の血中濃度が上昇するおそれがある。なお、重度の腎機能障害又は末期腎不全(クレアチニンクリアランス(CrCL):30mL/min以下)患者を対象とした有効性及び安全性を指標とした臨床試験は実施していない。[16.6.2参照]
9.3 肝機能障害患者
9.3.1 重度の肝機能障害のある患者
本剤の血中濃度が上昇するおそれがある。また、重度の肝機能障害(Child-Pugh分類C)患者を対象とした有効性及び安全性を指標とした臨床試験は実施していない。[16.6.1参照]
9.4 生殖能を有する者
9.4.1 妊娠する可能性のある女性には、本剤投与中及び最終投与後6カ月間において避妊する必要性及び適切な避妊法について説明すること。また、妊娠中に本剤を投与するか、本剤投与中の患者が妊娠した場合は、胎児に異常が生じる可能性があることを患者に十分説明すること。[9.5、15.2参照]
9.4.2 男性には、本剤投与中及び最終投与後3カ月間においてバリア法(コンドーム)を用いて避妊する必要性について説明すること。[15.2参照]
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。ラットを用いた動物実験において、臨床曝露量を下回る用量で胚・胎児死亡及び催奇形性(眼球異常、椎骨及び肋骨の欠損等)が報告されている。[9.4.1参照]
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。本剤の乳汁中への移行は不明である。
9.7 小児等
小児等を対象とした臨床試験は実施していない。
9.8 高齢者
患者の状態を観察しながら慎重に投与すること。一般に高齢者では、生理機能が低下している。
10. 相互作用
相互作用序文
本剤は、主にCYP3Aにより代謝される。[16.4参照]
薬物代謝酵素用語
CYP3A
10.2 併用注意
| 強いCYP3A阻害剤 イトラコナゾール リトナビル ボリコナゾール等 中程度のCYP3A阻害剤 シプロフロキサシン ジルチアゼム エリスロマイシン フルコナゾール ベラパミル等 [16.7.1参照] | 副作用の発現率及び重症度が増加するおそれがあるので、CYP3A阻害作用のない又は弱い薬剤への代替を考慮すること。やむを得ず中程度又は強いCYP3A阻害剤を併用する際には本剤の減量を考慮するとともに、患者の状態を慎重に観察し、副作用発現に十分注意すること。 | これらの薬剤等のCYP3A阻害作用により、本剤の代謝が阻害され、血中濃度が上昇する可能性がある。 |
| グレープフルーツ含有食品 | 本剤投与時はグレープフルーツ含有食品を摂取しないよう注意すること。 | これらの薬剤等のCYP3A阻害作用により、本剤の代謝が阻害され、血中濃度が上昇する可能性がある。 |
| CYP3A誘導剤 リファンピシン カルバマゼピン フェノバルビタール フェニトイン セイヨウオトギリソウ(St.John's Wort)含有食品等 [16.7.2参照] | 本剤の有効性が減弱するおそれがあるので、CYP3A誘導作用のない薬剤への代替を考慮すること。 | これらの薬剤等のCYP3A誘導作用により、本剤の代謝活性が誘導されるため、本剤の血中濃度が低下する可能性がある。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 骨髄抑制
貧血(30.2%)、好中球減少(13.7%)、白血球減少(12.0%)、リンパ球減少(7.0%)、血小板減少(6.8%)等があらわれることがある。[8.1参照]
11.1.2 間質性肺疾患(0.7%)
11.1.3 静脈血栓塞栓症
肺塞栓症(0.4%)、深部静脈血栓症(0.1%)等の静脈血栓塞栓症があらわれることがある。
11.1.4 感染症
肺炎(0.4%)等の重篤な感染症があらわれることがある。
11.1.5 赤芽球癆(1.6%)注)
本剤とデュルバルマブ(遺伝子組換え)との併用において、赤芽球癆があらわれることがある。
11.1.6 溶血性貧血(1.6%)注)
本剤とデュルバルマブ(遺伝子組換え)との併用において、溶血性貧血があらわれることがある。
11.1.7 肝機能障害(頻度不明)[8.1参照]
注)発現頻度は、国際共同第III相試験(DUO-E試験)における、本剤及びデュルバルマブ(遺伝子組換え)併用投与時から集計した。
11.2 その他の副作用
| 10%以上 | 1%〜10%未満 | 1%未満 | 頻度不明 | |
| 皮膚 | 発疹 | 過敏症、皮膚炎、結節性紅斑 | 血管性浮腫 | |
| 精神神経系 | 頭痛、浮動性めまい | |||
| 呼吸器 | 咳嗽、呼吸困難 | |||
| 消化器 | 悪心(47.4%)、嘔吐、下痢、食欲減退、味覚異常 | 消化不良、腹痛、便秘、口内炎、上腹部痛 | ||
| 全身 | 疲労・無力症(36.6%) | |||
| その他 | クレアチニン増加 | 平均赤血球容積(MCV)増加 |
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔を起こして縦隔洞炎等の重篤な合併症を併発することが報告されている。
15. その他の注意
15.1 臨床使用に基づく情報
国内外の臨床試験等において、骨髄異形成症候群、急性骨髄性白血病等の二次性悪性腫瘍が発生したとの報告がある。
15.2 非臨床試験に基づく情報
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
日本人固形癌患者(7例)に本剤300mgを単回経口投与したときのオラパリブの血漿中濃度推移及び薬物動態パラメータは以下のとおりであった2)。
図 日本人固形癌患者に本剤300mgを単回経口投与したときの血漿中オラパリブ濃度推移(算術平均値±標準偏差)
| 例数 | Cmax(μg/mL) | tmax(h)※ | AUC(μg・h/mL) | t1/2(h) |
| 7例 | 8.14±2.91 | 1.98(1.00〜3.00) | 54.4±37.5 | 9.43±2.86 |
16.1.2 反復投与
日本人固形癌患者(9例)に本剤200mg注1)及び300mgを1日2回反復経口投与したときの第15日目におけるオラパリブの血漿中濃度推移及び薬物動態パラメータは以下のとおりであった。また、300mg投与時におけるAUC(0-12時間)の累積係数は約1.8であった2)。
注1)本剤の承認用法・用量は300mgの1日2回投与である。
図 日本人固形癌患者に本剤200mg及び300mgを1日2回反復経口投与したときの第15日目における血漿中オラパリブ濃度推移(算術平均値±標準偏差)
| 用量 | 例数 | Cmax(μg/mL) | tmax(h)※ | AUC(0-12時間)(μg・h/mL) |
| 200mg | 3例 | 8.16±3.34 | 1.50(1.00〜3.00) | 41.1±20.9 |
| 300mg | 6例 | 8.86±3.14 | 3.00(1.50〜3.93) | 61.9±40.5 |
16.2 吸収
16.2.1 食事の影響
固形癌患者(56例)に本剤300mgを食後投与したとき、空腹時投与と比較して、オラパリブのCmaxは21%(90%信頼区間:14%〜28%)低下し、AUCは8%(90%信頼区間:1%〜16%)増加した3)(外国人データ)。
16.3 分布
オラパリブの血漿蛋白結合率はヒトでのCmax付近(10μg/mL)で82%であった。オラパリブの主要な結合蛋白は血清アルブミンであり(結合率:56%)、α1-酸性糖蛋白質との結合率は10μg/mLで29%であった4)(in vitro試験成績)。
16.4 代謝
In vitro試験から、オラパリブの主代謝酵素はCYP3A4/5であることが示された5)。
固形癌患者に14C標識オラパリブ100mgをカプセル剤注2)で単回経口投与したとき、投与12時間後までの血漿中において主成分はオラパリブであった(血漿中放射能の70%)。血漿中の主代謝物はM12(ピペラジン開環体の3位水酸化体)、M15(フルオロベンジル環のメチレン基水酸化体)及びM18(ピペラジン環の3位水酸化体)であった(血漿中放射能の9〜14%)。投与48時間後までの尿及び糞便中において主代謝物はM15であった(尿及び糞便中放射能のそれぞれ5〜6%)6)(外国人データ)。[10.参照]
固形癌患者に14C標識オラパリブ100mgをカプセル剤注2)で単回経口投与したとき、投与12時間後までの血漿中において主成分はオラパリブであった(血漿中放射能の70%)。血漿中の主代謝物はM12(ピペラジン開環体の3位水酸化体)、M15(フルオロベンジル環のメチレン基水酸化体)及びM18(ピペラジン環の3位水酸化体)であった(血漿中放射能の9〜14%)。投与48時間後までの尿及び糞便中において主代謝物はM15であった(尿及び糞便中放射能のそれぞれ5〜6%)6)(外国人データ)。[10.参照]
注2)カプセル剤は本邦未承認である。
16.5 排泄
固形癌患者に14C標識オラパリブ100mgをカプセル剤注3)で単回経口投与したとき、投与後7日間で投与放射能量の44%が尿中に、42%が糞便中に主に代謝物として排泄された。未変化体の尿中排泄率は15%であった6)(外国人データ)。
注3)カプセル剤は本邦未承認である。
16.6 特定の背景を有する患者
16.6.1 肝機能障害のある患者
肝機能の正常な固形癌患者並びに軽度(Child-Pugh分類A)又は中等度(Child-Pugh分類B)の肝機能障害を有する固形癌患者を対象に本剤300mgを単回経口投与した。軽度肝機能障害者(9例)では肝機能正常者(13例)に比べオラパリブのCmaxは13%(90%信頼区間:−18%〜56%)、AUCは15%(−28%〜83%)高値を示した。中等度肝機能障害者(8例)では肝機能正常者(13例)に比べオラパリブのCmaxは13%(90%信頼区間:−22%〜37%)低値を示したが、AUCは8%(−34%〜74%)高値を示した。軽度及び中等度の肝機能障害により臨床上問題となる影響は認められなかった7)(外国人データ)。[9.3.1参照]
16.6.2 腎機能障害のある患者
16.7 薬物相互作用
16.7.1 イトラコナゾール
固形癌患者(57例)に強いCYP3A阻害剤であるイトラコナゾール200mgを1日1回7日間投与し、投与5日目に本剤100mg注4)を併用投与したとき、オラパリブのCmaxは1.4倍(90%信頼区間:1.3〜1.5倍)に増加し、AUCは2.7倍(90%信頼区間:2.4〜3.0倍)に増加した9)(外国人データ)。また、生理学的薬物動態モデルによるシミュレーションから、本剤100mgと弱いCYP3A阻害剤であるフルボキサミンとの併用ではオラパリブのCmax及びAUC(0-t)に影響はないと推定されたものの、中程度のCYP3A阻害剤であるフルコナゾールとの併用ではオラパリブのCmax及びAUC(0-t)はそれぞれ平均1.14倍及び2.21倍増加すると推定された10)。[10.2参照]
注4)本剤の承認用法・用量は300mgの1日2回投与である。
16.7.2 リファンピシン
16.7.3 その他
17. 臨床成績
17.1 有効性及び安全性に関する試験
<白金系抗悪性腫瘍剤感受性の再発卵巣癌における維持療法>
17.1.1 国際共同第III相試験(SOLO2試験)16)
BRCA遺伝子変異陽性で、白金系抗悪性腫瘍剤を含む化学療法による少なくとも2回以上の治療歴があり、白金系抗悪性腫瘍剤感受性注1)かつ直近の白金系抗悪性腫瘍剤を含む化学療法で奏効(画像診断による完全奏効又は部分奏効)が維持されている再発高異型度漿液性卵巣癌(原発性腹膜癌及び卵管癌を含む)又は再発高異型度類内膜卵巣癌患者295例(本剤群196例、プラセボ群99例、うち日本人は本剤群8例、プラセボ群6例)を対象として、本剤(錠剤)300mg1日2回投与の有効性及び安全性をプラセボと比較する無作為化二重盲検プラセボ対照多施設共同第III相試験を実施した。主要評価項目である治験担当医師判定による無増悪生存期間において、本剤はプラセボに対し統計学的に有意な延長を示した(ハザード比0.30、95%信頼区間0.22〜0.41、p<0.0001)。無増悪生存期間の中央値は本剤群で19.1カ月、プラセボ群で5.5カ月であった。(2016年9月19日データカットオフ)
注1)PFI(platinum free interval)が6カ月以上であること。
図 SOLO2試験:無増悪生存期間のKaplan-Meier曲線(最大解析対象集団、治験担当医師による評価)
本剤が投与された195例(日本人8例を含む)中192例(98.5%)に有害事象が認められ、主な有害事象は、悪心148例(75.9%)、貧血84例(43.1%)、疲労74例(37.9%)、嘔吐73例(37.4%)、下痢64例(32.8%)、無力症61例(31.3%)であった。(2016年9月19日データカットオフ)
17.1.2 海外第II相試験(D0810C00019試験)17)
白金系抗悪性腫瘍剤を含む化学療法による少なくとも2回以上の治療歴があり、白金系抗悪性腫瘍剤感受性注2)かつ直近の白金系抗悪性腫瘍剤を含む化学療法で奏効(画像診断による完全奏効又は部分奏効)が維持されている再発漿液性卵巣癌(原発性腹膜癌及び卵管癌を含む)患者265例(本剤群136例、プラセボ群129例)を対象として、本剤(カプセル剤)400mg注3)1日2回投与の有効性及び安全性をプラセボと比較する無作為化二重盲検プラセボ対照多施設共同第II相試験を実施した。主要評価項目である治験担当医師判定による無増悪生存期間において、プラセボに対する本剤の優越性の評価で事前に設定した有効性判断基準を満たした(ハザード比0.35、95%信頼区間0.25〜0.49、p<0.00001[両側])。無増悪生存期間の中央値は本剤群で8.4カ月、プラセボ群で4.8カ月であった。(2010年6月30日データカットオフ)
注2)PFI(platinum free interval)が6カ月以上であること。
注3)本剤の承認用法・用量は錠剤300mgの1日2回投与である。
図 D0810C00019試験:無増悪生存期間のKaplan-Meier曲線(最大解析対象集団、治験担当医師による評価)
本剤が投与された136例中132例(97.1%)に有害事象が認められ、主な有害事象は、悪心96例(70.6%)、疲労73例(53.7%)、嘔吐48例(35.3%)であった。(2016年5月9日データカットオフ)
<BRCA遺伝子変異陽性の卵巣癌における初回化学療法後の維持療法>
17.1.3 国際共同第III相試験(SOLO1試験)18)
BRCA遺伝子変異陽性(病的変異又は病的変異疑い)で、新たに進行卵巣癌(FIGO進行期分類III期又はIV期)であると診断され、白金系抗悪性腫瘍剤を含む初回化学療法注4)で奏効(画像診断による完全奏効又は部分奏効)が維持されている高異型度漿液性又は高異型度類内膜卵巣癌(原発性腹膜癌及び卵管癌を含む)患者391例(本剤群260例、プラセボ群131例、うち日本人は本剤群11例、プラセボ群3例)を対象として、本剤(錠剤)300mg1日2回投与注5)の有効性及び安全性をプラセボと比較する無作為化二重盲検プラセボ対照多施設共同第III相試験を実施した。主要評価項目である治験担当医師判定による無増悪生存期間において、本剤はプラセボに対し統計学的に有意な延長を示した(ハザード比0.30、95%信頼区間0.23〜0.41、p<0.0001[両側])。無増悪生存期間の中央値は本剤群では未到達、プラセボ群で13.8カ月であった。(2018年5月17日データカットオフ)
注4)初回化学療法との併用又は初回化学療法後の維持療法としてベバシズマブ(遺伝子組換え)の投与を受けた患者は除外した。
注5)最長2年間又は原疾患の病勢進行が認められるまで投与した。投与開始2年後の時点で完全奏効(画像診断で病変なし)が維持されている場合は投与を中止し、投与開始2年後の時点で病変が確認され、治験担当医師が治療継続によりさらなるベネフィットが期待できると判断する場合は2年後以降も投与継続可能とした。
図 SOLO1試験:無増悪生存期間のKaplan-Meier曲線(最大解析対象集団、治験担当医師による評価)
本剤が投与された260例(日本人11例を含む)中256例(98.5%)に有害事象が認められ、主な有害事象は、悪心201例(77.3%)、疲労106例(40.8%)、嘔吐104例(40.0%)、貧血99例(38.1%)、下痢89例(34.2%)であった。(2018年5月17日データカットオフ)
<相同組換え修復欠損を有する卵巣癌におけるベバシズマブ(遺伝子組換え)を含む初回化学療法後の維持療法>
17.1.4 国際共同第III相試験(PAOLA-1試験)19)
新たに進行卵巣癌(FIGO進行期分類III期又はIV期)であると診断され、白金系抗悪性腫瘍剤、タキサン系抗悪性腫瘍剤及びベバシズマブによる初回化学療法注6)で奏効(画像診断による無病状態、完全奏効又は部分奏効)が維持されている高異型度漿液性又は類内膜卵巣癌(原発性腹膜癌及び卵管癌を含む)患者806例(本剤/ベバシズマブ群537例、プラセボ/ベバシズマブ群269例、うち日本人は本剤/ベバシズマブ群15例、プラセボ/ベバシズマブ群9例)を対象として、本剤(錠剤)300mg1日2回及びベバシズマブ併用投与注7)の有効性及び安全性をプラセボ及びベバシズマブ併用投与と比較する無作為化二重盲検プラセボ対照多施設共同第III相試験を実施した。主要評価項目である治験担当医師判定による無増悪生存期間において、本剤/ベバシズマブ群はプラセボ/ベバシズマブ群に対し統計学的に有意な延長を示した(ハザード比0.59、95%信頼区間0.49〜0.72、p<0.0001[両側])。(2019年3月22日データカットオフ)
また、腫瘍検体が入手可能であった755例のうち664例において相同組換え修復欠損に関する検査結果が得られ、探索的に実施された陽性・陰性注8)別の解析結果は下表のとおりであった。
また、腫瘍検体が入手可能であった755例のうち664例において相同組換え修復欠損に関する検査結果が得られ、探索的に実施された陽性・陰性注8)別の解析結果は下表のとおりであった。
注6)白金系抗悪性腫瘍剤とタキサン系抗悪性腫瘍剤との併用投与が6〜9サイクル行われ、少なくとも最後の3サイクルはベバシズマブも併用投与された患者を対象とした。
注7)最長2年間又は原疾患の病勢進行が認められるまで投与した。治験担当医師が治療継続によりさらなるベネフィットが期待できると判断する場合は2年後以降も投与継続可能とした。ベバシズマブは、無作為化前と無作為化後の合計で15カ月間まで投与可能とした。
注8)Myriad社の「Myriad myChoice HRD CDx」を用いた検査により、ゲノム不安定性スコア(GIS)が42以上、又はBRCA遺伝子変異陽性の場合に陽性と判定した。
| 相同組換え修復欠損 | 投与群 | 例数 | 中央値(カ月)[95%信頼区間] | ハザード比[95%信頼区間]※ |
| 陽性 | 本剤/ベバシズマブ群 | 255 | 37.2[36.0,−] | 0.33[0.25,0.45] |
| プラセボ/ベバシズマブ群 | 132 | 17.7[15.8,19.9] | ||
| 陰性 | 本剤/ベバシズマブ群 | 192 | 16.6[14.9,18.0] | 1.00[0.75,1.35] |
| プラセボ/ベバシズマブ群 | 85 | 16.2[13.8,18.6] |
図 PAOLA-1試験:無増悪生存期間のKaplan-Meier曲線(相同組換え修復欠損陽性集団、治験担当医師による評価)
本剤が投与された535例(日本人15例を含む)中531例(99.3%)に有害事象が認められ、主な有害事象は、悪心285例(53.3%)、疲労283例(52.9%)、高血圧245例(45.8%)、貧血219例(40.9%)であった。(2019年3月22日データカットオフ)
<がん化学療法歴のあるBRCA遺伝子変異陽性かつHER2陰性の手術不能又は再発乳癌>
17.1.5 国際共同第III相試験(OlympiAD試験)20)
生殖細胞系列のBRCA遺伝子変異(病的変異又は病的変異疑い)陽性かつHER2陰性であり、アントラサイクリン系抗悪性腫瘍剤(禁忌でない場合)及びタキサン系抗悪性腫瘍剤による治療歴を有する手術不能又は再発乳癌患者302例(本剤群205例、化学療法群97例、うち日本人は本剤群15例、化学療法群9例)を対象として、本剤300mg1日2回投与の有効性及び安全性を、医師が選択した化学療法(カペシタビン、エリブリン、又はビノレルビンのいずれかを選択)と比較する非盲検無作為化多施設共同第III相試験を実施した。主要評価項目である盲検下での独立中央評価に基づく無増悪生存期間において、本剤は医師が選択した化学療法に対し統計学的に有意な延長を示した(ハザード比0.58、95%信頼区間0.43〜0.80、p=0.0009[両側])。無増悪生存期間の中央値は本剤群で7.0カ月、化学療法群で4.2カ月であった。(2016年12月9日データカットオフ)
図 無増悪生存期間のKaplan-Meier曲線(OlympiAD試験:最大解析対象集団、盲検下での独立中央評価)
本剤が投与された205例(日本人15例を含む)中200例(97.6%)に有害事象が認められ、主な有害事象は、悪心119例(58.0%)、貧血81例(39.5%)、嘔吐66例(32.2%)であった。(2017年9月25日データカットオフ)
<BRCA遺伝子変異陽性かつHER2陰性で再発高リスクの乳癌における術後薬物療法>
17.1.6 国際共同第III相試験(OlympiA試験)21)
術前又は術後化学療法注9)歴のある生殖細胞系列のBRCA遺伝子変異(病的変異又は病的変異疑い)陽性かつHER2陰性で再発高リスク注10)の乳癌患者注11)1,836例(本剤群921例、プラセボ群915例、うち日本人は本剤群64例、プラセボ群76例)を対象として、本剤(錠剤)300mg1日2回投与の術後薬物療法としての有効性及び安全性を、プラセボと比較する無作為化二重盲検多施設共同第III相試験を実施した。本剤又はプラセボは最長1年間、又は疾患の再発若しくは許容できない毒性が認められるまで投与された。また、エストロゲン受容体(ER)及び/又はプロゲステロン受容体(PgR)陽性患者の場合、診療ガイドラインに従い、内分泌療法が併用された。主要評価項目である治験担当医師判定による浸潤性疾患のない生存期間(IDFS)において、本剤はプラセボに対して統計学的に有意な延長を示した(ハザード比0.581、95%信頼区間0.455〜0.737、p=0.0000073)。2年IDFS率は本剤群で89.2%、プラセボ群で81.5%であった。(2020年3月27日データカットオフ)
図 浸潤性疾患のない生存期間のKaplan-Meier曲線(OlympiA試験:最大解析対象集団、治験担当医師による評価)
本剤が投与された911例(日本人64例を含む)中836例(91.8%)に有害事象が認められ、主な有害事象は、悪心519例(57.0%)、疲労366例(40.2%)、貧血215例(23.6%)、嘔吐206例(22.6%)であった。(2021年7月12日データカットオフ)
注9)アントラサイクリン系抗悪性腫瘍剤若しくはタキサン系抗悪性腫瘍剤、又はその両剤を含む術前又は術後化学療法が少なくとも6サイクル実施された患者が対象とされた。
注10)以下の[1]又は[2]に該当する患者が再発高リスクの患者と定義された。
[1]術前化学療法歴のある場合
・乳房及び/又は切除リンパ節に残存浸潤性乳癌があるER及びPgR陰性かつHER2陰性乳癌患者(トリプルネガティブ乳癌患者)。
・乳房及び/又は切除リンパ節に残存浸潤性乳癌があり、CPS+EGスコア(臨床病期[CS]、ERの発現状態[E]、核グレード[G]及び治療後の病理学的病期[PS]−疾患スコア評価システム)が3以上であるER及び/又はPgR陽性かつHER2陰性乳癌患者。
[2]術後化学療法歴のある場合
・腋窩リンパ節転移陽性(≧pN1、腫瘍径は問わない)又は腋窩リンパ節転移陰性(pN0)であるが浸潤性原発腫瘍の病理学的サイズが2cmを超える(≧pT2)トリプルネガティブ乳癌患者。
・病理学的に確認した陽性リンパ節が4個以上あるER及び/又はPgR陽性かつHER2陰性乳癌患者。
注11)以下に該当する患者が対象とされた。
・乳房温存手術が施行された場合には、乳房に対する放射線療法が実施された患者。
・腋窩のセンチネルリンパ節転移陽性の場合には、腋窩リンパ節郭清術又は腋窩への放射線療法が実施された患者。
・放射線療法が実施された場合には、放射線療法終了後の患者。
<BRCA遺伝子変異陽性の遠隔転移を有する去勢抵抗性前立腺癌>
17.1.7 国際共同第III相試験(PROfound試験)22)
アビラテロン若しくはエンザルタミド又はその両剤の治療歴のある相同組換え修復関連遺伝子変異陽性注12)の遠隔転移を有する去勢抵抗性前立腺癌患者387例(本剤群256例、対照群131例、うち日本人は本剤群34例、対照群23例)を対象として、本剤(錠剤)300mg1日2回投与の有効性及び安全性を、治験担当医師が選択した治療(エンザルタミド又はアビラテロン酢酸エステル)と比較する無作為化非盲検多施設共同第III相試験を実施した。なお、両側精巣摘除術を受けていない患者は、性腺刺激ホルモン放出ホルモン(GnRH)アゴニスト又はアンタゴニスト療法が継続された。主要評価項目であるコホートAにおける盲検下での独立中央評価による画像診断に基づく無増悪生存期間において、本剤群は、対照群に対し統計学的に有意な延長を示した(ハザード比0.34、95%信頼区間0.25〜0.47、p<0.0001[両側])。(2019年6月4日データカットオフ)
また、探索的に実施されたBRCA1、BRCA2、ATM遺伝子変異別(該当する遺伝子変異のみが認められた患者集団)及びBRCA遺伝子変異陽性集団(少なくともBRCA1又はBRCA2遺伝子変異が認められた患者集団)の解析結果はそれぞれ下表[1]及び[2]のとおりであった。
また、探索的に実施されたBRCA1、BRCA2、ATM遺伝子変異別(該当する遺伝子変異のみが認められた患者集団)及びBRCA遺伝子変異陽性集団(少なくともBRCA1又はBRCA2遺伝子変異が認められた患者集団)の解析結果はそれぞれ下表[1]及び[2]のとおりであった。
注12)コホートAではBRCA1、BRCA2又はATM遺伝子に変異を有する患者、コホートBではBARD1、BRIP1、CDK12、CHEK1、CHEK2、FANCL、PALB2、PPP2R2A、RAD51B、RAD51C、RAD51D、又はRAD54L)に変異を有する患者を対象とした。
| 遺伝子変異 | 本剤群 | 対照群 | ハザード比[95%信頼区間]※ | ||
| 例数 | 中央値(カ月)[95%信頼区間] | 例数 | 中央値(カ月)[95%信頼区間] | ||
| BRCA1 | 8 | 2.07[1.38,5.52] | 5 | 1.84[1.71,3.71] | 0.41[0.13,1.39] |
| BRCA2 | 81 | 10.84[9.17,13.08] | 47 | 3.48[1.74,3.65] | 0.21[0.13,0.32] |
| ATM | 62 | 5.36[3.61,6.21] | 24 | 4.70[1.84,7.26] | 1.04[0.61,1.87] |
| 本剤群 | 対照群 | |
| 例数 | 102 | 58 |
| イベント数(%) | 62(60.8) | 51(87.9) |
| 中央値(カ月)[95%信頼区間] | 9.79[7.62,11.30] | 2.96[1.81,3.55] |
| ハザード比[95%信頼区間]※ | 0.22[0.15,0.32] | |
図 PROfound試験:画像診断に基づく無増悪生存期間のKaplan-Meier曲線(BRCA遺伝子変異陽性集団、盲検下での独立中央評価)
本剤が投与された256例(日本人34例を含む)中244例(95.3%)に有害事象が認められ、主な有害事象は、貧血118例(46.1%)、悪心106例(41.4%)、食欲減退77例(30.1%)であった。(2019年6月4日データカットオフ)
17.1.8 国際共同第III相試験(PROpel試験)23)
遠隔転移を有する去勢抵抗性前立腺癌(mCRPC)に対する薬物療法歴のない注13)mCRPC患者796例(本剤群399例、対照群397例、うち日本人は本剤群36例、対照群41例)を対象として、本剤300mg1日2回とアビラテロン酢酸エステル1000mg1日1回注14)併用投与の有効性及び安全性を、プラセボ1日2回とアビラテロン酢酸エステル1000mg1日1回併用投与と比較する無作為化二重盲検プラセボ対照多施設共同第III相試験を実施した。なお、両側精巣摘除術を受けていない患者は、性腺刺激ホルモン放出ホルモン(GnRH)アゴニスト又はアンタゴニスト療法が継続された。主要評価項目である治験担当医師の評価による画像診断に基づく無増悪生存期間(rPFS)において、本剤群は、プラセボ群に対して統計学的に有意な延長を示した(ハザード比0.66、95%信頼区間0.54〜0.81、p<0.0001[両側])。rPFSの中央値は本剤群で24.8カ月、プラセボ群で16.6カ月であった。(2021年7月30日データカットオフ)
また、探索的に実施されたBRCA遺伝子(BRCA1又はBRCA2遺伝子)変異の有無別のrPFS及び全生存期間(OS)の解析結果はそれぞれ下表のとおりであった。(2021年7月30日データカットオフ)
また、探索的に実施されたBRCA遺伝子(BRCA1又はBRCA2遺伝子)変異の有無別のrPFS及び全生存期間(OS)の解析結果はそれぞれ下表のとおりであった。(2021年7月30日データカットオフ)
注13)無作為割付け4週間前までのビカルタミド、フルタミド等の抗アンドロゲン剤の使用は許容された。また、mCRPCとなる前のエンザルタミド、アパルタミド、又はダロルタミドの使用については、それらの治療中に病勢進行がなく、無作為割付け12カ月間以上前に投与が終了している場合に限り許容された。限局性前立腺癌に対する術前・術後薬物療法中及び遠隔転移を有するホルモン感受性前立腺癌におけるドセタキセルの使用は、それらの治療中又は治療直後に治療無効又は病勢進行の徴候が認められていなければ許容された。
注14)アビラテロン酢酸エステルは、プレドニゾロン又はprednisone(国内未承認)(いずれも5mg1日2回経口投与)と併用された。
| 遺伝子変異 | 投与群 | 例数 | 中央値(カ月)[95%信頼区間] | ハザード比※1[95%信頼区間] | |
| rPFS | BRCA陽性※2 | 本剤群 | 47 | −[−,−] | 0.23[0.12,0.43] |
| プラセボ群 | 38 | 8.4[5.5,14.8] | |||
| BRCA陰性※3 | 本剤群 | 214 | 21.9[16.6,25.2] | 0.86[0.66,1.12] | |
| プラセボ群 | 213 | 16.7[13.8,19.4] | |||
| OS | BRCA陽性※2 | 本剤群 | 47 | −[−,−] | 0.39[0.16,0.86] |
| プラセボ群 | 38 | 23.6[17.8,−] | |||
| BRCA陰性※3 | 本剤群 | 214 | −[−,−] | 1.10[0.78,1.57] | |
| プラセボ群 | 213 | −[−,−] |
図 PROpel試験:rPFSのKaplan-Meier曲線(腫瘍組織検体又は血漿検体の少なくとも一方におけるBRCA遺伝子変異陽性集団、治験担当医師による評価)
本剤が投与された398例(日本人36例を含む)中387例(97.2%)に有害事象が認められ、主な有害事象は、貧血181例(45.5%)、悪心112例(28.1%)、疲労111例(27.9%)であった。(2021年7月30日データカットオフ)
<BRCA遺伝子変異陽性の治癒切除不能な膵癌における白金系抗悪性腫瘍剤を含む化学療法後の維持療法>
17.1.9 海外第III相試験(POLO試験)24)
生殖細胞系列のBRCA遺伝子変異陽性(病的変異又は病的変異疑い)で、白金系抗悪性腫瘍剤を含む一次化学療法が16週間以上継続された後、疾患進行が認められていない遠隔転移を有する膵腺癌患者154例(本剤群92例、プラセボ群62例)を対象として、本剤(錠剤)300mg1日2回投与の維持療法における有効性及び安全性をプラセボと比較する無作為化二重盲検プラセボ対照多施設共同第III相試験を実施した。主要評価項目である盲検下での独立中央評価による無増悪生存期間において、本剤はプラセボと比較して統計学的に有意な延長を示した(ハザード比0.53、95%信頼区間0.35〜0.82、p=0.0038[両側]、中央値:本剤群7.4カ月、プラセボ群3.8カ月)。(2019年1月15日データカットオフ)
図 POLO試験:無増悪生存期間のKaplan-Meier曲線(最大解析対象集団、盲検下での独立中央評価)
本剤が投与された91例中87例(95.6%)に有害事象が認められ、主な有害事象は、疲労41例(45.1%)、悪心41例(45.1%)であった。(2019年1月15日データカットオフ)
<ミスマッチ修復機能正常(pMMR)の進行・再発の子宮体癌におけるデュルバルマブ(遺伝子組換え)を含む化学療法後の維持療法>
17.1.10 国際共同第III相試験(DUO-E試験)25)
化学療法歴のない注15)進行・再発注16)の子宮体癌患者注17)718例([1]本剤/デュルバルマブ/化学療法群注18)239例、[2]デュルバルマブ/化学療法群注18)238例、[3]化学療法群注18)241例、うち、日本人はそれぞれ[1]26例、[2]30例、[3]32例)を対象として、上記[1]及び[2]の有効性及び安全性を、[3]と比較する無作為化二重盲検プラセボ対照多施設共同第III相試験を実施した。
主要評価項目である治験担当医師判定による無増悪生存期間において、本剤/デュルバルマブ/化学療法群及びデュルバルマブ/化学療法群は化学療法群に対して統計学的に有意な延長を示した([1]化学療法群に対する本剤/デュルバルマブ/化学療法群のハザード比0.55、95%信頼区間0.43〜0.69、p<0.0001[両側]、中央値:本剤/デュルバルマブ/化学療法群15.1カ月、化学療法群9.6カ月、[2]化学療法群に対するデュルバルマブ/化学療法群のハザード比0.71、95%信頼区間0.57〜0.89、p=0.003[両側]、中央値:デュルバルマブ/化学療法群10.2カ月、化学療法群9.6カ月)。(2023年4月12日データカットオフ)
pMMRの患者集団及びミスマッチ修復機能欠損(dMMR)の患者集団における無増悪生存期間はそれぞれ下表のとおりであった。
主要評価項目である治験担当医師判定による無増悪生存期間において、本剤/デュルバルマブ/化学療法群及びデュルバルマブ/化学療法群は化学療法群に対して統計学的に有意な延長を示した([1]化学療法群に対する本剤/デュルバルマブ/化学療法群のハザード比0.55、95%信頼区間0.43〜0.69、p<0.0001[両側]、中央値:本剤/デュルバルマブ/化学療法群15.1カ月、化学療法群9.6カ月、[2]化学療法群に対するデュルバルマブ/化学療法群のハザード比0.71、95%信頼区間0.57〜0.89、p=0.003[両側]、中央値:デュルバルマブ/化学療法群10.2カ月、化学療法群9.6カ月)。(2023年4月12日データカットオフ)
pMMRの患者集団及びミスマッチ修復機能欠損(dMMR)の患者集団における無増悪生存期間はそれぞれ下表のとおりであった。
注15)術前又は術後の補助療法として抗悪性腫瘍剤が投与された場合には、抗悪性腫瘍剤の最終投与日から再発までの期間が12カ月間以上の患者が対象とされた。
注16)以下のいずれかに該当する患者が対象とされた。
・International Federation of Gynecology and Obstetrics(FIGO)分類(2009年版)III期のうち、手術又は生検後にRECIST ver.1.1に基づく測定可能病変が認められた患者
・FIGO分類(2009年版)IV期の患者(手術又は生検後の残存病変の有無は問わない)
・手術を含む治療により根治する可能性が低い再発の患者
注17)組織型は問わず、病理組織学的に上皮性子宮体癌と診断された患者が対象とされた(癌肉腫は組入れ可能とされ、子宮肉腫は組入れ不可とされた)。
注18)用法・用量は以下のとおりとされた。
| 投与群 | 化学療法併用期 | 維持療法期 |
| 本剤/デュルバルマブ/化学療法群 | CBDCA及びPTX†1、2との併用で、デュルバルマブ1120mgをQ3Wで静脈内投与†3 | ・デュルバルマブ1500mgをQ4Wで静脈内投与 ・本剤300mgをBID経口投与 |
| デュルバルマブ/化学療法群 | CBDCA及びPTX†1、2との併用で、デュルバルマブ1120mgをQ3Wで静脈内投与†3 | ・デュルバルマブ1500mgをQ4Wで静脈内投与 ・本剤のプラセボをBID経口投与 |
| 化学療法群 | CBDCA及びPTX†1、2との併用で、デュルバルマブのプラセボをQ3Wで静脈内投与†3 | ・デュルバルマブのプラセボをQ4Wで静脈内投与 ・本剤のプラセボをBID経口投与 |
| 本剤/デュルバルマブ/化学療法群 | デュルバルマブ/化学療法群 | 化学療法群 | |
| 例数 | 191 | 192 | 192 |
| 中央値[95%信頼区間](カ月) | 15.0[12.4,18.0] | 9.9[9.4,12.5] | 9.7[9.2,10.1] |
| ハザード比[95%信頼区間]*1,*3 | 0.57[0.44,0.73] | 0.77[0.60,0.97] | 該当なし |
| ハザード比[95%信頼区間]*2,*3 | 0.76[0.59,0.99] | 該当なし | 該当なし |
| 本剤/デュルバルマブ/化学療法群 | デュルバルマブ/化学療法群 | 化学療法群 | |
| 例数 | 48 | 46 | 49 |
| 中央値[95%信頼区間](カ月) | 31.8[12.4,未達] | 未達[未達,未達] | 7.0[6.7,14.8] |
| ハザード比[95%信頼区間]*1,*3 | 0.41[0.21,0.75] | 0.42[0.22,0.80] | 該当なし |
| ハザード比[95%信頼区間]*2,*3 | 0.97[0.49,1.98] | 該当なし | 該当なし |
図 DUO-E試験:無増悪生存期間のKaplan-Meier曲線(pMMRの患者集団、治験担当医師判定)
本剤が投与された192例(日本人20例を含む)中184例(95.8%)に有害事象が認められ、主な有害事象は、悪心79例(41.1%)、貧血70例(36.5%)、疲労43例(22.4%)及び嘔吐39例(20.3%)であった。(2023年4月12日データカットオフ)
18. 薬効薬理
18.1 作用機序
19. 有効成分に関する理化学的知見
19.1. オラパリブ
| 一般的名称 | オラパリブ |
|---|---|
| 一般的名称(欧名) | Olaparib |
| 化学名 | 4-[(3-{[4-(Cyclopropylcarbonyl)piperazin-1-yl]carbonyl}-4-fluorophenyl)methyl]phthalazin-1(2H)-one |
| 分子式 | C24H23FN4O3 |
| 分子量 | 434.46 |
| 物理化学的性状 | 本品は白色〜微黄色の粉末である。 |
| KEGG DRUG |
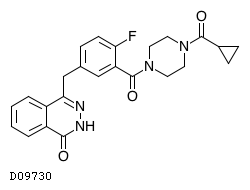
|
20. 取扱い上の注意
防湿のためPTP包装のまま保存すること。
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
<リムパーザ錠100mg>
56錠[8錠(PTP)×7]
<リムパーザ錠150mg>
56錠[8錠(PTP)×7]
23. 主要文献
- 社内資料(遺伝毒性試験)(2018年1月19日承認、CTD2.6.6.4), (2017)
- 社内資料(日本人固形癌患者における薬物動態)(2018年1月19日承認、CTD2.7.6.2.4.2.4、2.7.2.3.1.6), (2014)
- Ruth Plummer R,et al., Cancer Chemother Pharmacol., 76 (4), 723-729, (2015) »PubMed
- 社内資料(血漿蛋白結合[in vitro試験])(2018年1月19日承認、CTD2.7.2.3.1.2、2.7.2.4.2.1.1、2.7.2.4.2.1.2、2.7.2.4.2.1.3), (2017)
- 社内資料(代謝に関与する代謝酵素[in vitro試験])(2018年1月19日承認、CTD2.6.4.5.3), (2010)
- 社内資料(ヒトに[14C]-オラパリブを投与したマスバランス試験)(2018年1月19日承認、CTD2.7.2.3.1.3、2.7.2.3.1.4、2.6.4.5.4.2.4、2.6.5.9.3), (2009)
- 社内資料(肝機能障害を有する固形癌患者における薬物動態)(2018年7月2日承認、CTD2.7.2.3.2.3), (2016)
- 社内資料(腎機能障害を有する固形癌患者における薬物動態)(2018年1月19日承認、CTD2.7.2.3.2.4), (2015)
- Dirix L,et al., Clin Ther., 38 (10), 2286-2299, (2016) »PubMed
- 社内資料(CYPに対する阻害作用[in vitro試験])(2018年1月19日承認、CTD2.7.2.3.3.2.2、2.7.2.2.3.1), (2014)
- 社内資料(CYPに対する誘導作用[in vitro試験])(2018年1月19日承認、CTD2.7.2.3.3.2.2、2.7.2.2.3.1), (2015)
- 社内資料(UGTに対する阻害作用[in vitro試験])(2020年12月25日承認、CTD2.6.4.7.1、2.6.5.15.1), (2019)
- 社内資料(内分泌療法剤の相互作用)(2018年1月19日承認、CTD2.7.2.2.2.10、2.7.6.2.6.2.3), (2015)
- 社内資料(P-糖蛋白質の関与)(2018年1月19日承認、CTD2.7.2.4.2.4.1), (2007)
- 社内資料(トランスポーターに対する阻害作用)(2018年1月19日承認、CTD2.7.2.3.3.2.2), (2014)
- Pujade-Lauraine E,et al., Lancet Oncol., 18, 1274-1284, (2017) »PubMed
- Ledermann J,et al., N Engl J Med., 366, 1382-1392, (2012) »PubMed
- Moore K,et al., N Engl J Med., 379, 2495-2505, (2018) »PubMed
- Ray-Coquard I,et al., N Engl J Med., 381, 2416-2428, (2019) »PubMed
- Robson M,et al., N Engl J Med., 377, 523-533, (2017) »PubMed
- Tutt ANJ,et al., N Engl J Med., 384, 2394-2405, (2021) »PubMed
- de Bono J,et al., N Engl J Med., 382, 2091-2102, (2020) »PubMed
- Clarke NW,et al., N Engl J Med Evid., 1 (9), (2022), (doi:10.1056/EVIDoa2200043)
- Golan T,et al., N Engl J Med., 381, 317-327, (2019) »PubMed
- Westin SN,et al., J Clin Oncol., 42 (3), 283-299, (2024) »PubMed
- Menear KA,et al., J Med Chem., 51, 6581-6591, (2008) »PubMed
- 社内資料(各種腫瘍細胞株の増殖に対するオラパリブの作用[in vitro試験])(2018年1月19日承認、CTD2.6.2.2.3), (2013)
- 社内資料(HBCx-10腫瘍移植モデルにおけるオラパリブのPK、PD及び有効性の評価[in vivo試験])(2018年1月19日承認、CTD2.6.2.2.5), (2016)
24. 文献請求先及び問い合わせ先
文献請求先
アストラゼネカ株式会社
メディカルインフォメーションセンター
〒530-0011
大阪市北区大深町3番1号
電話:0120-189-115
製品情報問い合わせ先
アストラゼネカ株式会社
メディカルインフォメーションセンター
〒530-0011
大阪市北区大深町3番1号
電話:0120-189-115
26. 製造販売業者等
26.1 製造販売元
アストラゼネカ株式会社
大阪市北区大深町3番1号
26.2 プロモーション提携
MSD株式会社
東京都千代田区九段北1-13-12