医薬品情報
| 総称名 | プレバイミス |
|---|---|
| 一般名 | レテルモビル |
| 欧文一般名 | Letermovir |
| 製剤名 | レテルモビル注射液 |
| 薬効分類名 | 抗サイトメガロウイルス化学療法剤 |
| 薬効分類番号 | 6250 |
| ATCコード | J05AX18 |
| KEGG DRUG |
D10801
レテルモビル
|
| KEGG DGROUP |
DG01937
抗サイトメガロウイルス薬
|
| JAPIC | 添付文書(PDF) |
添付文書情報2025年10月 改訂(第6版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| プレバイミス点滴静注240mg | PREVYMIS Intravenous Infusion 240mg | MSD | 6250406A1029 | 20962円/瓶 | 劇薬, 処方箋医薬品 |
1. 警告
<同種造血幹細胞移植>
同種造血幹細胞移植患者の感染管理に十分な知識・経験を持つ医師のもとで、本剤の投与が適切と判断される症例のみに投与すること。
2. 禁忌
次の患者には投与しないこと
2.1 本剤の成分に対し過敏症の既往歴のある患者
2.2 次の薬剤を投与中の患者
ピモジド、エルゴタミン酒石酸塩・無水カフェイン・イソプロピルアンチピリン、ジヒドロエルゴタミン、メチルエルゴメトリン、エルゴメトリン[10.1参照]
4. 効能または効果
下記におけるサイトメガロウイルス感染症の発症抑制
○同種造血幹細胞移植
○臓器移植
5. 効能または効果に関連する注意
<臓器移植>
腎移植以外の臓器移植患者を対象に本剤の有効性及び安全性を評価する臨床試験は実施していない。
6. 用法及び用量
通常、成人にはレテルモビルとして480mgを1日1回、約60分かけて点滴静注する。シクロスポリンと併用投与する場合にはレテルモビルとして240mgを1日1回、約60分かけて点滴静注する。
通常、小児にはレテルモビルとして以下の用量を1日1回、約60分かけて点滴静注する。
| 体重 | 用量 (シクロスポリンの併用なし) | 用量 (シクロスポリンの併用あり) |
| 30kg以上 | 480mg | 240mg |
| 体重 | 用量 (シクロスポリンの併用の有無にかかわらない) | |
| 15kg以上30kg未満 | 120mg | |
| 7.5kg以上15kg未満 | 60mg | |
| 5kg以上7.5kg未満 | 40mg | |
7. 用法及び用量に関連する注意
<効能共通>
7.1 経口剤(錠剤及び顆粒剤)と注射剤は医師の判断で切り替えて使用することができる。なお、体重30kg未満の小児では、切り替える際に用量の調節が必要となる場合がある。ただし、臨床試験において注射剤の長期投与の経験はなく、注射剤の添加剤ヒドロキシプロピル-β-シクロデキストリンは腎機能障害のある患者で蓄積し、腎機能の悪化等を引き起こすおそれがあることから、注射剤の投与は最小限の期間とし、経口投与可能な患者には、経口投与を選択すること。[9.2.1参照]
<同種造血幹細胞移植>
<臓器移植>
8. 重要な基本的注意
長期間に亘り点滴静注製剤を継続して使用する場合には、添加剤ヒドロキシプロピル-β-シクロデキストリンの蓄積により腎機能障害の悪化等を引き起こすおそれがあるため、定期的に腎機能検査を実施する等観察を十分に行うこと。[15.2.2参照]
9. 特定の背景を有する患者に関する注意
9.2 腎機能障害患者
9.2.1 中等度又は重度(クレアチニンクリアランス<50mL/min)の腎機能障害のある患者
定期的に腎機能検査を実施する等観察を十分に行うこと。添加剤ヒドロキシプロピル-β-シクロデキストリンの蓄積により腎機能障害の悪化等を引き起こすおそれがある。[15.2.2参照]
9.3 肝機能障害患者
9.4 生殖能を有する者
妊娠可能な女性に対しては、本剤が胎児に悪影響を及ぼす可能性があることを十分に説明し、本剤投与中及び本剤投与終了後一定期間は適切な避妊を行うよう指導すること。[9.5参照]
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、本剤投与の有益性が危険性を上回ると判断される場合にのみ投与すること。妊娠中に本剤を投与するか、本剤投与中の患者が妊娠した場合は、本剤投与による催奇形性等が生じる可能性があることについて、患者に十分説明すること。
妊娠ラット及びウサギの器官形成期に投与したとき、成人同種造血幹細胞移植患者の臨床曝露量(480mg静脈内投与)のそれぞれ11倍及び1.7倍の母動物毒性を示す用量で骨格奇形、胎児体重の減少等が認められた。妊娠ラットに着床から分娩後まで投与した試験では、臨床曝露量の2.2倍まで胚・胎児毒性は認められなかった。[9.4参照]
妊娠ラット及びウサギの器官形成期に投与したとき、成人同種造血幹細胞移植患者の臨床曝露量(480mg静脈内投与)のそれぞれ11倍及び1.7倍の母動物毒性を示す用量で骨格奇形、胎児体重の減少等が認められた。妊娠ラットに着床から分娩後まで投与した試験では、臨床曝露量の2.2倍まで胚・胎児毒性は認められなかった。[9.4参照]
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。動物試験(ラット)で乳汁移行が認められている1)。
9.7 小児等
<臓器移植>
小児等を対象とした臨床試験は実施していない。
10. 相互作用
相互作用序文
レテルモビルは有機アニオン輸送ポリペプチド1B1/3(OATP1B1/3)、P-糖蛋白(P-gp)及びUDP-グルクロノシルトランスフェラーゼ1A1/3(UGT1A1/3)の基質である。レテルモビルはCYP3Aの時間依存的な阻害作用、並びに乳癌耐性蛋白(BCRP)及びOATP1B1/3の阻害作用を有する。また、レテルモビルはCYP2C9及びCYP2C19の誘導作用を有する可能性がある。[16.7.1参照]
薬物代謝酵素用語
有機アニオン輸送ポリペプチド1B1(OATP1B1)
薬物代謝酵素用語
有機アニオン輸送ポリペプチド1B3(OATP1B3)
薬物代謝酵素用語
P-糖蛋白(P-gp)
薬物代謝酵素用語
UDP-グルクロノシルトランスフェラーゼ1A1(UGT1A1)
薬物代謝酵素用語
UDP-グルクロノシルトランスフェラーゼ1A3(UGT1A3)
薬物代謝酵素用語
CYP3A
薬物代謝酵素用語
乳癌耐性蛋白(BCRP)
薬物代謝酵素用語
CYP2C9
薬物代謝酵素用語
CYP2C19
10.1 併用禁忌
10.2 併用注意
| CYP3Aの基質 フェンタニル キニジン ミダゾラム等 [16.7.2参照] | 併用により、これらの薬剤の血漿中濃度が上昇するおそれがある。 | レテルモビルの併用により、CYP3Aが阻害されると予測される。 |
| ボリコナゾール [16.7.2参照] | 併用により、ボリコナゾールの血漿中濃度が低下する。 併用時は、ボリコナゾールの治療効果を減弱させるおそれがあるため、患者の状態を十分に観察することが推奨される。 | レテルモビルの併用により、CYP2C9及びCYP2C19が誘導されると考えられる。 |
| CYP2C9又はCYP2C19の基質 フェニトイン ワルファリン等 | 併用により、これらの薬剤の血漿中濃度が低下するおそれがある。 フェニトインとの併用時は、血中フェニトイン濃度を頻繁にモニタリングすること。 ワルファリンとの併用時は、INRを頻繁にモニタリングすること。 | レテルモビルの併用により、CYP2C9又はCYP2C19が誘導されると予測される。 |
| リファンピシン [16.7.2参照] | 併用により、レテルモビルの血漿中濃度が低下する。 また、リファンピシンとの併用終了翌日に単独投与したレテルモビルの血漿中濃度がさらに低下するので、リファンピシンとの併用終了後、レテルモビルの有効性が減弱する可能性がある。 | リファンピシンの併用により、P-gp及びUGT1A1/3が誘導されると考えられる。 |
| アトルバスタチン [16.7.2参照] | 併用により、アトルバスタチンの血漿中濃度が上昇する。 併用時は、アトルバスタチンの副作用(ミオパチー等)に注意して患者の状態を十分に観察すること。 | レテルモビルの併用により、CYP3A、OATP1B1/3及び腸管のBCRPが阻害される。 |
| シンバスタチン | 併用により、これらの薬剤の血漿中濃度が上昇するおそれがある。 併用時は、これらの薬剤の副作用(ミオパチー等)に注意して患者の状態を十分に観察すること。 | レテルモビルの併用により、CYP3A、OATP1B1/3及び腸管のBCRPが阻害されると予測される。 |
| ロスバスタチン フルバスタチン | 併用により、これらの薬剤の血漿中濃度が上昇するおそれがある。 併用時は、これらの薬剤の副作用(ミオパチー等)に注意して患者の状態を十分に観察すること。 | レテルモビルの併用により、OATP1B1/3及び腸管のBCRPが阻害されると予測される。 |
| プラバスタチン ピタバスタチン | 併用により、これらの薬剤の血漿中濃度が上昇するおそれがある。 併用時は、これらの薬剤の副作用(ミオパチー等)に注意して患者の状態を十分に観察すること。 | レテルモビルの併用により、OATP1B1/3が阻害されると予測される。 |
| シクロスポリン [16.7.2参照] | 併用により、レテルモビル及びシクロスポリンの血中濃度が上昇する。 レテルモビルとの併用時及び中止時には、シクロスポリンの血中濃度を頻繁にモニタリングし、シクロスポリンの用量を調節すること。 | レテルモビルの併用により、CYP3Aが阻害される。 シクロスポリンの併用により、OATP1B1/3が阻害される。 |
| タクロリムス シロリムス [16.7.2参照] | 併用により、これらの薬剤の血中濃度が上昇する。 レテルモビルとの併用時及び中止時には、これらの薬剤の血中濃度を頻繁にモニタリングし、これらの薬剤の用量を調節すること。 | レテルモビルの併用により、CYP3Aが阻害される。 |
| エベロリムス [16.7.3参照] | 併用により、エベロリムスの血中濃度が上昇するおそれがある。 レテルモビルとの併用時及び中止時には、エベロリムスの血中濃度を頻繁にモニタリングし、エベロリムスの用量を調節すること。 | レテルモビルの併用により、CYP3Aが阻害されると予測される。 |
11. 副作用
11.2 その他の副作用
| 1%以上5%未満 | 1%未満 | |
| 血液及びリンパ系障害 | 白血球減少症 | 好中球減少症 |
| 胃腸障害 | 悪心、下痢、嘔吐 | |
| 免疫系障害 | 過敏症 | |
| 臨床検査 | 白血球数減少 |
14. 適用上の注意
14.1 薬剤調製時の注意
14.1.1 希釈前に、変色や不溶性異物がないか、各バイアルを確認すること。本剤は無色澄明の溶液である。また、製品由来の少量の半透明又は白色の微粒子を含むことがある。バイアル内の溶液に変色や濁り、又は異物(少量の半透明又は白色の微粒子以外)が認められた場合は使用しないこと。バイアルを振盪しないこと。
14.1.2
バイアルから溶液を採取して日局生理食塩液又は日局5%ブドウ糖注射液が入った点滴バッグに添加し、振盪せず静かに混和すること。
60〜480mgを投与する場合、バイアルからの採取液量、点滴バッグの液量は以下を参照し、点滴バッグの総液量を投与すること。
60〜480mgを投与する場合、バイアルからの採取液量、点滴バッグの液量は以下を参照し、点滴バッグの総液量を投与すること。
| レテルモビルの投与量 | バイアルからの採取液量 | 点滴バッグの液量 |
| 480mg | 24mL(12mL×2バイアル) | 250mL |
| 240mg | 12mL | 250mL |
| 120mg | 6mL | 75mL |
| 60mg | 3mL | 50mL |
40mgを投与する場合、1バイアル(レテルモビル濃度20mg/mL)から5mLを採取し、45mLの日局生理食塩液又は日局5%ブドウ糖注射液が入った点滴バッグに添加し、振盪せず静かに混和すること。当該希釈液を20mL投与すること。
14.1.3 本剤のバイアルは1回使い切りである。残液は使用しないこと。
14.1.4 混和後、本剤の希釈液は無色〜黄色澄明の溶液となる。投与前の希釈液に変色や不溶性異物がないか目視により確認すること。変色や濁り、又は異物(少量の半透明又は白色の微粒子以外)が認められる場合には、希釈液を廃棄すること。
14.1.5 希釈液は、室温保存(2〜30℃)では24時間以内に、冷蔵保存(2〜8℃)した場合は48時間以内に使用すること。なお、これらの時間には点滴終了までの時間が含まれる。
14.2 配合変化
本剤は他剤と配合したとき、濁りや不溶性異物が生じることがある。配合適性についてはデータが限られているが、次の薬剤は配合禁忌であり、同一の輸液ラインを通して同時に注入しないこと。
主な配合禁忌薬剤
アミオダロン塩酸塩、アムホテリシンBリポソーム、アズトレオナム、セフェピム塩酸塩、シプロフロキサシン、シクロスポリン、ジルチアゼム塩酸塩、フィルグラスチム(遺伝子組換え)、ゲンタマイシン硫酸塩、レボフロキサシン、リネゾリド、ロラゼパム、ミダゾラム、オンダンセトロン塩酸塩、パロノセトロン塩酸塩
14.3 薬剤投与時の注意
14.3.1 必ず0.2μmインラインフィルター(ポリエーテルスルホン、ポリスルホン又は正荷電ナイロン製)を使用して投与すること。
14.3.2 本剤はポリウレタンを含有する輸液チューブで投与しないこと。
15. その他の注意
15.2 非臨床試験に基づく情報
15.2.1 動物試験(ラット)において、成人同種造血幹細胞移植患者の臨床曝露量(480mg静脈内投与)の3倍以上の曝露量で精巣毒性(精細管の変性、精子数の低値、精子の運動性低下、異常精子発現率の増加、受胎能への影響等)が認められた。ラット精巣毒性に対する無毒性量での曝露量は、臨床曝露量と同程度であった。雄マウス及びサルでは、動物における最高用量(臨床曝露量のそれぞれ3.5倍及び2.1倍)まで精巣への影響は認められなかった。第III相試験ではレテルモビルに関連した精巣毒性を示唆する所見は認められなかった。
15.2.2 添加剤であるヒドロキシプロピル-β-シクロデキストリンをラット及びイヌへ静脈内投与すると50mg/kgを超える用量で腎臓及び膀胱の空胞化等の生理学的な適応性変化を引き起こすことが報告されている2)。
16. 薬物動態
16.1 血中濃度
16.1.1 健康成人
日本人健康成人女性にレテルモビル240mg注)及び480mgを60分かけて単回静脈内投与した際の、レテルモビルの薬物動態パラメータを表1に示す。レテルモビルは、二相性の消失を示した。また、レテルモビルのAUC0-∞は、用量比を上回る上昇を示した。
| 用量 | 例数 | Ceoi†(ng/mL) | AUC0-∞(ng・hr/mL) | t1/2(hr) |
| 240mg注) | 6 | 18,700(16.2) | 60,800(20.2) | 11.8(64.0) |
| 480mg | 6 | 41,000(21.3) | 176,000(31.9) | 10.8(33.7) |
また、日本人健康成人女性にレテルモビル480mgを反復経口投与した際、AUC0-24hr及びCmaxの幾何平均比に基づく累積係数は、それぞれ0.97及び0.94であった。
16.1.2 成人同種造血幹細胞移植患者
成人同種造血幹細胞移植患者350例(うち、日本人成人同種造血幹細胞移植患者23例)から得られた血漿中レテルモビル濃度データを用いて、母集団薬物動態解析を実施した。日本人成人同種造血幹細胞移植患者にレテルモビルを480mg、シクロスポリンを併用投与する場合はレテルモビルを240mgで1日1回静脈内投与した際の、レテルモビルの定常状態におけるAUC0-24hrを表2に示す。第III相国際共同試験(001試験)で得られた曝露量の範囲では、一貫した有効性が示されており、各投与方法における曝露量に、臨床的な違いは認められなかった。
| 投与方法 | AUC0-24hr†(ng・hr/mL) | ||
| 例数 | 幾何平均 | 幾何平均に基づく変動係数(%) | |
| 480mg静脈内投与 | 11 | 101,200 | 24.4 |
| シクロスポリン併用240mg静脈内投与 | 6 | 70,810 | 16.5 |
16.1.3 小児同種造血幹細胞移植患者
小児同種造血幹細胞移植患者60例(うち、日本人小児同種造血幹細胞移植患者4例)から得られた血漿中レテルモビル濃度データを用いて、母集団薬物動態解析を実施した。小児同種造血幹細胞移植患者にレテルモビルを1日1回静脈内投与した際の、レテルモビルの定常状態におけるAUC0-24hrを表3に示す。小児同種造血幹細胞移植患者の曝露量は、すべての体重区分で、成人同種造血幹細胞移植患者で得られた曝露量の範囲内であった。
| 体重 | シクロスポリン非併用時の静脈内投与量 | AUC0-24hr(ng・hr/mL)中央値 [90%予測区間]† | シクロスポリン併用時の静脈内投与量 | AUC0-24hr(ng・hr/mL)中央値 [90%予測区間]† |
| 30kg以上 | 480mg | 111,000 [55,700,218,000] | 240mg | 59,800 [28,400,120,000] |
| 15kg以上30kg未満 | 120mg | 57,200 [29,700,113,000] | 120mg | 61,100 [29,900,121,000] |
| 7.5kg以上15kg未満 | 60mg | 46,000 [24,300,83,900] | 60mg | 49,200 [25,800,93,800] |
| 5kg以上7.5kg未満 | 40mg | 43,400 [24,300,81,000] | 40mg | 45,900 [24,900,82,200] |
16.3 分布
母集団薬物動態解析から、日本人を含む成人同種造血幹細胞移植患者にレテルモビルを静脈内投与した際の、レテルモビルの定常状態における分布容積の平均値は、45.5Lと推定された。
In vitroデータより、レテルモビルは、ヒト血漿蛋白に対し、高い結合を示した(98.7%)。レテルモビルの血中と血漿中濃度比(血中/血漿)は0.56であり、検討した濃度範囲(0.1〜10mg/L)で変わらなかった。
非臨床分布試験から、レテルモビルは、消化管、胆管及び肝臓の臓器並びに組織に高濃度に分布し、脳に低濃度に分布した。
16.4 代謝
外国人健康成人に、ラベル体で標識したレテルモビルを経口投与した際、血漿中レテルモビル関連物質の大部分は未変化体であり(96.6%)、主要代謝物は検出されなかった。レテルモビルは、UGT1A1/1A3を介したグルクロン酸抱合により、一部消失した。
16.5 排泄
母集団薬物動態解析から、日本人を含む成人同種造血幹細胞移植患者にレテルモビルを静脈内投与した際、レテルモビルの定常状態におけるクリアランスは、4.84L/hrと推定された。また、クリアランスの個体間変動は、24.6%と推定された。外国人健康成人に、ラベル体で標識したレテルモビルを経口投与した際、総放射能の93.3%は糞中から回収された。レテルモビルは主に未変化体として糞中に排泄され、少量(6%)はアシルグルクロン酸抱合体として排泄された。また、レテルモビルの腎排泄は、わずかであった(2%未満)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害者
外国人成人腎機能障害者を対象とした臨床試験で、レテルモビルを1日1回8日間反復経口投与した際、成人腎機能正常者(推算糸球体濾過量が90mL/min/1.73m2以上)と比較して、レテルモビルのAUC0-24hrは、中等度(推算糸球体濾過量が30〜59mL/min/1.73m2)の成人腎機能障害者では約1.9倍及び重度(推算糸球体濾過量が30mL/min/1.73m2未満)の成人腎機能障害者では約1.4倍高かった。
外国人成人腎移植患者における母集団薬物動態解析から、軽度(クレアチニンクリアランスが60mL/min以上90mL/min未満)、中等度(クレアチニンクリアランスが30mL/min以上60mL/min未満)及び重度(クレアチニンクリアランスが15mL/min以上30mL/min未満)の腎機能障害を有する成人被験者におけるレテルモビルのAUCは、クレアチニンクリアランスが90mL/min以上の成人被験者と比較してそれぞれ約1.1倍、1.3倍及び1.4倍高かったが、臨床的に意味はないと考えられた。
16.6.2 肝機能障害者
16.7 薬物相互作用
16.7.1 In vitro試験
In vitroデータから、レテルモビルは、OATP1B1/3、P-gp、BCRP、UGT1A1及びUGT1A3の基質であることが示唆された。また、レテルモビルは、CYP3Aの時間依存的な阻害作用又は誘導作用、CYP2C8の可逆的な阻害作用、CYP2B6の誘導作用を有することが示唆された。また、レテルモビルは、排出トランスポーターであるP-gp、BCRP、胆汁酸塩輸送ポンプ(BSEP)、多剤耐性関連蛋白(MRP2)、有機アニオントランスポーター(OAT3)及び肝取り込みトランスポーターであるOATP1B1/3の阻害作用を有することが示唆された。
16.7.2 臨床薬物相互作用試験
健康成人を対象とした臨床薬物相互作用試験から得られた、レテルモビルの薬物動態に及ぼす併用薬の影響及び併用薬の薬物動態に及ぼすレテルモビルの影響についてそれぞれ表4及び表5に示す。[10.2参照]
| 併用薬 | 併用薬の投与方法 | レテルモビルの投与方法 | 例数 | レテルモビルの薬物動態パラメータの幾何平均比 (併用時/非併用時) (90%信頼区間) | |
| AUC | Cmax | ||||
| 抗真菌薬 | |||||
| フルコナゾール | 400mg 単回 PO | 480mg 単回 PO | 14 | 1.11 (1.01,1.23) | 1.06 (0.93,1.21) |
| イトラコナゾール | 200mg QD PO | 480mg QD PO | 14 | 1.33 (1.17,1.51) | 1.21 (1.05,1.39) |
| 抗マイコバクテリア薬 | |||||
| リファンピシン | 600mg 単回 PO | 480mg 単回 PO | 16 | 2.03 (1.84,2.26) | 1.59 (1.46,1.74) |
| 600mg 単回 IV | 480mg 単回 PO | 16 | 1.58 (1.38,1.81) | 1.37 (1.16,1.61) | |
| 600mg QD PO† | 480mg QD PO | 14 | 0.81 (0.67,0.98) | 1.01 (0.79,1.28) | |
| 600mg QD PO (リファンピシン併用終了後24時間)†† | 480mg QD PO | 14 | 0.15 (0.13,0.17) | 0.27 (0.22,0.31) | |
| 免疫抑制薬 | |||||
| シクロスポリン§ | 200mg 単回 PO | 240mg QD PO | 12 | 2.11 (1.97,2.26) | 1.48 (1.33,1.65) |
| ミコフェノール酸モフェチル | 1g 単回 PO | 480mg QD PO | 14 | 1.18 (1.04,1.32) | 1.11 (0.92,1.34) |
| タクロリムス | 5mg 単回 PO | 80mg BID PO注) | 14 | 1.02 (0.97,1.07) | 0.92 (0.84,1.00) |
| 併用薬 | 併用薬の投与方法 | レテルモビルの投与方法 | 例数 | 併用薬の薬物動態パラメータの幾何平均比 (併用時/非併用時) (90%信頼区間) | |
| AUC | Cmax | ||||
| CYP3A基質 | |||||
| ミダゾラム | 1mg 単回 IV | 240mg QD PO注) | 16 | 1.47 (1.37,1.58) | 1.05 (0.94,1.17) |
| 2mg 単回 PO | 240mg QD PO注) | 16 | 2.25 (2.04,2.48)† | 1.72 (1.55,1.92) | |
| P-gp基質 | |||||
| ジゴキシン | 0.5mg 単回 PO | 240mg BID PO注) | 22 | 0.88 (0.80,0.96)† | 0.75 (0.63,0.89) |
| 免疫抑制薬 | |||||
| シクロスポリン | 50mg 単回 PO | 240mg QD PO | 14 | 1.66 (1.51,1.82) | 1.08 (0.97,1.19) |
| ミコフェノール酸モフェチル | 1g 単回 PO | 480mg QD PO | 14 | 1.08 (0.97,1.20) | 0.96 (0.82,1.12) |
| タクロリムス | 5mg 単回 PO | 480mg QD PO | 13 | 2.42 (2.04,2.88) | 1.57 (1.32,1.86) |
| シロリムス | 2mg 単回 PO | 480mg QD PO | 13 | 3.40 (3.01,3.85) | 2.76 (2.48,3.06) |
| 抗真菌薬及び抗ウイルス薬 | |||||
| アシクロビル | 400mg 単回 PO | 480mg QD PO | 13 | 1.02 (0.87,1.20) | 0.82 (0.71,0.93) |
| フルコナゾール | 400mg 単回 PO | 480mg 単回 PO | 14 | 1.03 (0.99,1.08) | 0.95 (0.92,0.99) |
| イトラコナゾール | 200mg QD PO | 480mg QD PO | 14 | 0.76 (0.71,0.81) | 0.84 (0.76,0.92) |
| ポサコナゾール | 300mg 単回 PO | 480mg QD PO | 13 | 0.98 (0.82,1.17) | 1.11 (0.95,1.29) |
| ボリコナゾール | 200mg BID PO | 480mg QD PO | 12 | 0.56 (0.51,0.62) | 0.61 (0.53,0.71) |
| HMG-CoA還元酵素阻害剤 | |||||
| アトルバスタチン | 20mg 単回 PO | 480mg QD PO | 14 | 3.29 (2.84,3.82) | 2.17 (1.76,2.67) |
| 経口避妊薬 | |||||
| エチニルエストラジオール/レボノルゲストレル | 0.03mg EE 単回 PO | 480mg QD PO | 22 | 1.42 (1.32,1.52) | 0.89 (0.83,0.96) |
| 0.15mg LNG 単回 PO | 22 | 1.36 (1.30,1.43) | 0.95 (0.86,1.04) | ||
16.7.3 生理学的薬物速度論モデルによるシミュレーション
注)本剤の用法・用量は、成人にはレテルモビルとして1日1回480mgを静脈内投与である。なお、シクロスポリンを併用投与する場合には、1日1回240mgを静脈内投与である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
<同種造血幹細胞移植>
17.1.1 第III相国際共同試験(001試験)
日本人を含むCMV抗体陽性の成人同種造血幹細胞移植患者(無作為化された患者570例、うち日本人患者36例)を対象に、CMV感染症の発症抑制効果及び安全性を検討することを目的として、プラセボ対照無作為化二重盲検並行群間比較試験(001試験)を実施した。移植日から移植後28日までの期間にレテルモビル480mg(シクロスポリン併用時はレテルモビル240mg)又はプラセボの投与を開始し、1日1回、経口又は静脈内投与にて、移植後14週(約100日)まで投与した。主要評価項目である移植後24週以内に臨床的に意味のあるCMV感染注1)が認められた被験者の割合は、レテルモビル群とプラセボ群の比較において、統計学的に有意な差が認められた6)。
注1)臨床的に意味のあるCMV感染
CMV血症の確認及び被験者の臨床状態に基づく抗CMV薬による先制治療の開始、又は臓器障害を伴うCMV感染症の発症
| レテルモビル群 (325例) | プラセボ群 (170例) | |
| 移植後24週以内に臨床的に意味のあるCMV感染が認められた被験者の割合† | 37.5% (122/325例) | 60.6% (103/170例) |
| プラセボとの群間差 [95.02%信頼区間]†† | −23.5 [−32.6,−14.5] | − |
| p値†† | <0.0001 | − |
17.1.2 第III相国際共同試験(040試験)
移植後14週(約100日)以降もCMV感染及び感染症リスクを有するCMV抗体陽性の成人同種造血幹細胞移植患者注2)(無作為化された患者220例、うち日本人患者16例)を対象に、レテルモビルの予防投与を移植後14週(約100日)から移植後28週(約200日)に延長した際の有効性及び安全性を検討することを目的として、プラセボ対照無作為化二重盲検並行群間比較試験(040試験)を実施した。移植後約100日までレテルモビルによる予防投与を完了した患者を無作為に割り付け、レテルモビル480mg(シクロスポリン併用時はレテルモビル240mg)又はプラセボを、1日1回、経口又は静脈内投与にて、移植後28週(約200日)まで投与した。主要評価項目である移植後14週(約100日)から28週(約200日)までに臨床的に意味のあるCMV感染注1)が認められた被験者の割合は、レテルモビル群とプラセボ群の比較において、統計学的に有意な差が認められた7)。
注1)臨床的に意味のあるCMV感染
CMV血症の確認及び被験者の臨床状態に基づく抗CMV薬による先制治療の開始、又は臓器障害を伴うCMV感染症の発症
注2)移植後14週(約100日)以降もCMV感染及び感染症リスクを有する患者として、次の基準を1つ以上満たした者が含まれた
血縁ドナーで3つのHLA遺伝子座(HLA-A、HLA-B又はHLA-DR)の少なくとも1つに1箇所以上の不一致がある、HLA半合致ドナー、非血縁ドナーで4つのHLA遺伝子座(HLA-A、HLA-B、HLA-C及びHLA-DRB1)の少なくとも1つに1箇所以上の不一致がある、臍帯血移植、ex vivo T細胞除去移植、抗ヒト胸腺細胞免疫グロブリン使用、アレムツズマブ使用、無作為割付け前6週以内に1mg/kg/日以上のプレドニゾロン(又は同等の薬剤)の全身投与
| レテルモビル(レテルモビル約200日投与)群 (144例) | プラセボ(レテルモビル約100日投与)群 (74例) | |
| 移植後14週から28週に臨床的に意味のあるCMV感染が認められた被験者の割合† | 2.8% (4/144例) | 18.9% (14/74例) |
| プラセボとの群間差 [95.02%信頼区間]†† | −16.1 [−25.8,−6.4] | − |
| p値†† | 0.0005 | − |
17.1.3 後期第II相国際共同試験(030試験)
CMV感染及び感染症リスクを有する出生時から18歳未満の小児同種造血幹細胞移植患者(組み入れられた患者65例、うち日本人患者5例)を対象に、レテルモビルを投与した際の薬物動態、CMV感染症の発症抑制効果及び安全性を検討することを目的として、単群非盲検試験(030試験)を実施した。移植日から移植後28日までの期間に年齢、体重及び剤形に基づく用量のレテルモビルの投与を開始し、1日1回、経口又は静脈内投与にて、移植後14週(約100日)まで投与した。有効性評価項目は、移植後24週以内の臨床的に意味のあるCMV感染注1)であった。移植後24週以内に臨床的に意味のあるCMV感染が認められた被験者の割合注3)は、25.0%(14/56例)であった。
注1)臨床的に意味のあるCMV感染
CMV血症の確認及び被験者の臨床状態に基づく抗CMV薬による先制治療の開始、又は臓器障害を伴うCMV感染症の発症
注3)移植後24週以内の治験中止例又は移植後24週時点の有効性データの欠測例は不成功例とした。
<臓器移植>
17.1.4 第III相海外試験(002試験)
CMV抗体陽性のドナーより移植を受けるCMV抗体陰性の外国人成人腎移植患者(無作為化された患者601例)を対象に、CMV感染症の発症抑制効果及び安全性を検討することを目的として、無作為化二重盲検実薬対照非劣性試験(002試験)を実施した。移植日から移植後7日までの期間にレテルモビル480mg注4)(シクロスポリン併用時はレテルモビル240mg)又はバルガンシクロビル900mg(静脈内投与の場合はガンシクロビル5mg/kg)の投与を開始し、1日1回、経口又は静脈内投与にて、移植後28週(約200日)まで投与した。レテルモビル群の被験者には単純ヘルペスウイルス及び水痘帯状疱疹ウイルスの予防のためアシクロビルを投与し、バルガンシクロビル群の被験者にはアシクロビルのプラセボを投与した。主要評価項目である移植後52週以内にCMV感染症を発症した被験者の割合は表3のとおりであった。10%の非劣性マージンに基づき、レテルモビルはバルガンシクロビルに対して非劣性を示した8)。
注4)シクロスポリン非併用時に静脈内投与する場合は、レテルモビル240mg又は480mg投与のいずれかに割り付けられる試験デザインであったが、240mgで投与された被験者は1例であった。本剤の承認用量は480mgである(シクロスポリン非併用時)。
| レテルモビル群 (289例) | バルガンシクロビル群 (297例) | |
| 移植後52週以内にCMV感染症を発症した被験者†の割合†† | 10.4% (30/289例) | 11.8% (35/297例) |
| バルガンシクロビル群との群間差 [95%信頼区間]§ | −1.4 [−6.5,3.8] | − |
17.1.5 第III相国内試験(042試験)
ドナー又はレシピエントいずれかのCMV抗体が陽性の日本人成人腎移植患者(22例)を対象に、CMV感染症の発症抑制効果及び安全性を検討することを目的として、単群非盲検試験(042試験)を実施した。移植日から移植後7日までの期間にレテルモビル480mg(シクロスポリン併用時はレテルモビル240mg)の投与を開始し、1日1回、経口投与にて、移植後28週(約200日)まで投与した。移植後52週以内にCMV感染症を発症した被験者(独立した中央判定委員会で臓器障害を伴うCMV感染症又はCMV症候群と判定された被験者)の割合は、9.5%(2/21例)であった。
移植後28週までに、レテルモビルの投与を受けた22例中4例(18.2%)に副作用が認められた。報告された副作用は白血球減少症、下痢、悪心及び血中アルカリホスファターゼ増加(各1例、4.5%)であった9)。[7.2、7.4参照]
移植後28週までに、レテルモビルの投与を受けた22例中4例(18.2%)に副作用が認められた。報告された副作用は白血球減少症、下痢、悪心及び血中アルカリホスファターゼ増加(各1例、4.5%)であった9)。[7.2、7.4参照]
17.3 その他
17.3.1 心電図に及ぼす影響
TQT試験で、外国人健康成人38例を対象に、レテルモビルがQTc間隔に及ぼす影響をプラセボ及び陽性対照と比較検討した。レテルモビル960mgを単回静脈内投与注5)したときのQTcP間隔(試験集団固有のべき係数で補正したQT間隔)のベースラインからの変化量のプラセボとの差[90%信頼区間]の最大値は、4.93[2.81,7.05]ms(投与後1時間)であった。
注5)本剤の用法・用量は、成人にはレテルモビルとして1日1回480mgを静脈内投与である。なお、シクロスポリンを併用投与する場合には、1日1回240mgを静脈内投与である。
18. 薬効薬理
18.1 作用機序
レテルモビルはウイルスの複製に必要なCMVのDNAターミナーゼ複合体を阻害する。生化学的な検討及び電子顕微鏡所見から、レテルモビルは一単位長のゲノムの生成に影響し、ウイルス粒子の形成を阻害することが明らかとなった。
18.2 In vitro抗ウイルス作用
感染細胞培養系でのCMVの臨床分離株(74株)に対するレテルモビルのEC50値の範囲は0.7〜6.1nMであった。
18.3 耐性ウイルス
18.3.1 細胞培養系
CMVのDNAターミナーゼのサブユニットはCMV遺伝子のUL51、UL56及びUL89領域にコードされる。細胞培養系にてレテルモビルに低感受性のCMV変異株を分離した。その結果、pUL51(P91S、A95V)、pUL56(C25F、S229F、V231A/L、N232Y、V236A/L/M、E237D、L241P、T244K/R、L254F、L257F/I、K258E、F261C/L/S、Y321C、C325F/R/W/Y、L328V、M329T、A365S、N368D、R369G/M/S)及びpUL89(N320H、D344E)にアミノ酸置換が認められた。これらの置換を有する遺伝子組換えCMV変異株のEC50値は野生株と比較して1.6〜9,300倍高値を示した。
18.3.2 臨床試験
外国人を対象とした第II相試験(020試験)では、131例の成人同種造血幹細胞移植患者に60、120又は240mgのレテルモビル又はプラセボを1日1回84日間投与し、レテルモビル群のうち予防不成功となり検体が得られた12例を対象に、UL56遺伝子の231〜369位のアミノ酸配列を中心にDNAシークエンス解析を実施した。60mg投与群1例でレテルモビルに低感受性を示す置換(V236M)が検出された。
成人同種造血幹細胞移植患者を対象とした第III相国際共同試験(001試験)では、レテルモビル群のうち予防不成功となり検体が得られた50例を対象に、UL56及びUL89遺伝子のすべてのコード領域のDNAシークエンス解析を実施した。3例でレテルモビルに低感受性を示す4種類の置換がpUL56に検出された。1例でC325W及びR369Tが、他の2例で各々V236M及びE237Gの置換が検出された。
成人同種造血幹細胞移植患者を対象とした第III相国際共同試験(040試験)では、全投与群のうち予防不成功又は早期中止しCMV血症が認められた32例を対象に、UL51、UL56及びUL89遺伝子のすべてのコード領域のDNAシークエンス解析を実施した。レテルモビルに低感受性を示す置換は検出されなかった。
小児同種造血幹細胞移植患者を対象とした後期第II相国際共同試験(030試験)では、CMV血症が認められ検体が得られた10例を対象に、UL51、UL56及びUL89遺伝子のすべてのコード領域のDNAシークエンス解析を実施した。2例でレテルモビルに低感受性を示す置換がpUL56に検出された。1例でR369Sが、他の1例でC325Wの置換が検出された。
外国人成人腎移植患者を対象とした第III相海外試験(002試験)では、レテルモビル群のうちCMV感染症を発症又は早期中止しCMV血症が認められ検体が得られた52例を対象に、UL51、UL56及びUL89遺伝子のすべてのコード領域のDNAシークエンス解析を実施した。レテルモビルに低感受性を示す置換は検出されなかった。
日本人成人腎移植患者を対象とした第III相国内試験(042試験)では、レテルモビルの投与を受けた被験者のうちCMV感染症を発症又はCMV血症が認められ検体が得られた4例を対象に、UL51、UL56及びUL89遺伝子のすべてのコード領域のDNAシークエンス解析を実施した。レテルモビルに低感受性を示す置換は検出されなかった。
18.4 交差耐性
ガンシクロビルに耐性を示すpUL97又はpUL54領域に置換を有するCMVは、レテルモビルに感受性を示した。野生型と比較してガンシクロビルに対する感受性を2.1倍低下させるpUL56 E237G置換を有する遺伝子組換えCMV株を除き、レテルモビルに対し耐性を示す置換を有する各種遺伝子組換え株は、ホスカルネット及びガンシクロビルに対して感受性を示した。
19. 有効成分に関する理化学的知見
19.1. レテルモビル
| 一般的名称 | レテルモビル |
|---|---|
| 一般的名称(欧名) | Letermovir |
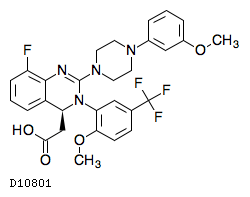
| 化学名 | (4S)-2-{8-Fluoro-2-[4-(3-methoxyphenyl)piperazin-1-yl]-3-[2-methoxy-5-(trifluoromethyl)phenyl]-3,4-dihydroquinazolin-4-yl}acetic acid |
| 分子式 | C29H28F4N4O4 |
| 分子量 | 572.55 |
| 物理化学的性状 | 白色の粉末である。 |
| KEGG DRUG |
|
20. 取扱い上の注意
外箱開封後は遮光して保存すること。
21. 承認条件
21.1 医薬品リスク管理計画を策定の上、適切に実施すること。
21.2 腎以外の臓器移植患者を対象とした臨床試験は実施されていないことから、製造販売後、一定数の症例に係るデータが集積されるまでの間は、腎以外の臓器移植患者については全症例を対象に使用成績調査を実施することにより、本剤の使用患者の背景情報を把握するとともに、本剤の安全性及び有効性に関するデータを早期に収集し、本剤の適正使用に必要な措置を講じること。
22. 包装
1バイアル(12mL)×10
23. 主要文献
- 社内資料:レテルモビルのラット乳汁中移行に関する試験(2018年3月23日承認、CTD2.6.4.6)
- Gould S,et al., Food Chem Toxicol., 43, 1451-9, (2005) »PubMed
- Crumling MA,et al., Front Cell Neurosci., 11, 355, (2017) »PubMed
- Liu X,et al., Neurotox Res., 38, 808-23, (2020) »PubMed
- Menzel K,et al., Clin Transl Sci., 16, 1039-48, (2023) »PubMed
- Marty FM,et al., N Engl J Med., 377, 2433-44, (2017) »PubMed
- Russo D,et al., Lancet Haematol., 11, e127-35, (2024)
- Limaye AP,et al., JAMA., 330, 33-42, (2023) »PubMed
- Ishida H,et al., Clin Exp Nephrol., 28, 822-31, (2024) »PubMed
24. 文献請求先及び問い合わせ先
文献請求先
MSD株式会社
MSDカスタマーサポートセンター
東京都千代田区九段北1-13-12
電話:医療関係者の方:フリーダイヤル 0120-024-961
製品情報問い合わせ先
MSD株式会社
MSDカスタマーサポートセンター
東京都千代田区九段北1-13-12
電話:医療関係者の方:フリーダイヤル 0120-024-961
26. 製造販売業者等
26.1 製造販売元
MSD株式会社
東京都千代田区九段北1-13-12