医薬品情報
| 総称名 | ビバンセ |
|---|---|
| 一般名 | リスデキサンフェタミンメシル酸塩 |
| 欧文一般名 | Lisdexamfetamine Mesilate |
| 製剤名 | リスデキサンフェタミンメシル酸塩カプセル |
| 薬効分類名 | 中枢神経刺激剤 |
| 薬効分類番号 | 1179 |
| ATCコード | N06BA12 |
| KEGG DRUG |
D04747
リスデキサンフェタミンメシル酸塩
|
| JAPIC | 添付文書(PDF) |
添付文書情報2025年11月 改訂(第4版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ビバンセカプセル20mg | Vyvanse Capsules | 武田薬品工業 | 1179059M1024 | 660.5円/カプセル | 劇薬, 覚醒剤原料, 処方箋医薬品注) |
| ビバンセカプセル30mg | Vyvanse Capsules | 武田薬品工業 | 1179059M2020 | 731.4円/カプセル | 劇薬, 覚醒剤原料, 処方箋医薬品注) |
1. 警告
1.1 本剤の投与は、注意欠陥/多動性障害(AD/HD)の診断、治療に精通し、かつ薬物依存を含む本剤のリスク等についても十分に管理できる、管理システムに登録された医師のいる医療機関及び薬剤師のいる薬局において、登録患者に対してのみ行うこと。また、それら薬局においては、調剤前に当該医師・医療機関・患者が登録されていることを確認した上で調剤を行うこと。
1.2 本剤の投与にあたっては、患者又は代諾者に対して、本剤の有効性、安全性、及び目的以外への使用や他人への譲渡をしないことを文書によって説明し、文書で同意を取得すること。
2. 禁忌
次の患者には投与しないこと
2.1 本剤の成分又は交感神経刺激アミン(メタンフェタミン、メチルフェニデート、ノルアドレナリン、アドレナリン、ドパミン等)に対し過敏症の既往歴のある患者
2.2 重篤な心血管障害のある患者[血圧又は心拍数を上昇させ、症状を悪化させるおそれがある。][8.5参照]
2.3 甲状腺機能亢進のある患者[循環器系に影響を及ぼすことがある。]
2.4 過度の不安、緊張、興奮性のある患者[中枢神経刺激作用により症状を悪化させることがある。]
2.5 運動性チックのある患者、Tourette症候群又はその既往歴・家族歴のある患者[症状を悪化又は誘発させることがある。]
2.6 薬物乱用の既往歴のある患者[慢性的乱用により過度の耐性及び様々な程度の異常行動を伴う精神的依存を生じるおそれがある。]
2.7 閉塞隅角緑内障のある患者[眼圧を上昇させるおそれがある。]
2.8 褐色細胞腫又はパラガングリオーマのある患者[血圧を上昇させるおそれがある。]
2.9 モノアミン酸化酵素(MAO)阻害剤(セレギリン塩酸塩、ラサギリンメシル酸塩、サフィナミドメシル酸塩)を投与中又は投与中止後2週間以内の患者[高血圧クリーゼに至るおそれがある。][10.1参照]
4. 効能または効果
小児期における注意欠陥/多動性障害(AD/HD)
5. 効能または効果に関連する注意
5.1 本剤の使用実態下における乱用・依存性に関する評価が行われるまでの間は、他のAD/HD治療薬が効果不十分な場合にのみ使用すること。
5.3 本剤による薬物治療を18歳未満で開始した患者において、18歳以降も継続して本剤を投与する場合には、治療上の有益性と危険性を考慮して慎重に投与するとともに、定期的に本剤の有効性及び安全性を評価し、有用性が認められない場合には、投与中止を考慮し、漫然と投与しないこと。
5.4 AD/HDの診断は、米国精神医学会の精神疾患の診断・統計マニュアル(DSM※)等の標準的で確立した診断基準に基づき慎重に実施し、基準を満たす場合にのみ投与すること。
※:Diagnostic and Statistical Manual of Mental Disorders
6. 用法及び用量
通常、小児にはリスデキサンフェタミンメシル酸塩として30mgを1日1回朝経口投与する。症状により、1日70mgを超えない範囲で適宜増減するが、増量は1週間以上の間隔をあけて1日用量として20mgを超えない範囲で行うこと。
7. 用法及び用量に関連する注意
7.1 本剤の投与量は必要最小限となるよう、患者ごとに慎重に観察しながら調節すること。
7.2 高度の腎機能障害のある患者(GFR30mL/min/1.73m2未満)には、1日用量として50mgを超えて投与しないこと。また、透析患者又はGFR15mL/min/1.73m2未満の患者では、更に低用量の投与を考慮し、増量に際しては患者の状態を十分に観察すること。[9.2.1、13.2、16.6.1参照]
7.3 不眠があらわれるおそれがあるため、就寝時間等を考慮し、午後の服用は避けること。
8. 重要な基本的注意
8.1 本剤を投与する医師又は医療従事者は、投与前に患者及び保護者又はそれに代わる適切な者に対して、本剤の治療上の位置づけ、依存性等を含む本剤のリスクについて、十分な情報を提供するとともに、適切な使用方法について指導すること。
8.2 本剤を長期間投与する場合には、個々の患者に対して定期的に休薬期間を設定して有用性の再評価を実施すること。
8.3 まれに視覚障害の症状(調節障害、霧視)が報告されている。視覚障害が認められた場合には、眼科検査を実施し、必要に応じて投与を中断又は中止すること。
8.4 めまい、眠気、視覚障害等が起こることがあるので、本剤投与中の患者には自動車の運転等危険を伴う機械の操作には従事させないよう注意すること。
8.5 本剤の国内外臨床試験において0〜16.7%に血圧上昇(20mmHg以上)、7.4〜26.5%に脈拍数増加(20bpm以上)が認められた。本剤は血圧又は心拍数に影響を与えることがあるので、本剤投与開始前及び投与期間中は以下の点に注意すること。[2.2、9.1.1参照]
8.5.1 心血管系に対する影響を観察するため、本剤投与開始前及び投与期間中は、定期的に心拍数(脈拍数)及び血圧を測定すること。
8.5.2 本剤を心血管障害のある患者に投与する際は、循環器を専門とする医師に相談するなど、慎重に投与の可否を検討すること。
8.5.3 患者の心疾患に関する病歴、突然死や重篤な心疾患に関する家族歴等から、心臓に重篤ではないが異常が認められる、又はそのおそれがある患者に対して本剤の投与を検討する場合には、投与開始前に心電図検査等により心血管系の状態を評価すること。また、本剤投与中に労作性胸痛、原因不明の失神、又は他の心疾患を示唆する症状を示した場合は、直ちに心血管系の状態を評価し、本剤の投与を中止するなど適切な処置を行うこと。
8.6 双極性障害の患者ではうつ状態から混合状態/躁状態に移行するおそれがあることから、うつ症状のある患者に対して本剤の投与を検討する場合には、患者の精神系疾患歴、自殺、双極性障害及びうつ病の家族歴等から双極性障害の可能性がないか評価すること。[9.1.2参照]
8.7 通常量の本剤を服用していた精神病性障害の既往がない患者において、幻覚、妄想等の症状が報告されている。これらの症状があらわれた場合には本剤の投与を中止すること。
8.8 通常量の本剤を服用していた精神病性障害や躁病の既往がない患者において、躁病等が報告されている。これらの症状があらわれた場合には本剤との関連の可能性を考慮し、必要に応じて減量又は投与を中止するなど適切な処置を行うこと。
8.9 自殺念慮や自殺行為があらわれることがあるので、患者の状態を注意深く観察すること。また、患者及び保護者又はそれに代わる適切な者に対し、これらの症状・行為があらわれた場合には、速やかに医療機関に連絡するよう指導すること。
8.10 攻撃性、敵意はAD/HDにおいてしばしば観察されるが、本剤の投与中にも攻撃性、敵意の発現や悪化が報告されている。投与中は、攻撃的行動、敵意の発現又は悪化について観察すること。
8.11 本剤の投与により体重増加の抑制、成長遅延が報告されている。本剤の投与中は患児の成長に注意し、身長や体重の増加が思わしくないときは、投与の中断等を考慮すること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 高血圧又は不整脈のある患者
血圧又は心拍数の上昇により症状を悪化させるおそれがある。[8.5参照]
9.1.2 精神系疾患(精神病性障害、双極性障害)のある患者
行動障害、思考障害又は躁病エピソードの症状が悪化するおそれがある。[8.6参照]
9.1.3 痙攣発作、脳波異常又はその既往歴のある患者
痙攣閾値を低下させ、発作を誘発するおそれがある。
9.1.4 脳血管障害(脳動脈瘤、血管炎、脳卒中等)又はその既往歴のある患者
症状を悪化又は再発させるおそれがある。
9.2 腎機能障害患者
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。出生前又は出生後早期に、本剤の活性体であるアンフェタミンの臨床用量相当量を曝露したげっ歯類において、出生児に学習障害、記憶障害若しくは自発運動量の変化等の長期の神経行動学的変化、発育遅延又は生殖能への影響が認められている。
9.6 授乳婦
授乳しないことが望ましい。ヒト母乳中へ移行することが報告されている。
9.7 小児等
10. 相互作用
10.1 併用禁忌
| モノアミン酸化酵素(MAO)阻害剤 セレギリン塩酸塩 (エフピー) ラサギリンメシル酸塩 (アジレクト) サフィナミドメシル酸塩 (エクフィナ) [2.9参照] | MAO阻害剤を投与中あるいは投与中止後2週間以内の患者には本剤を投与しないこと。高血圧クリーゼが起こるおそれがある。また、死亡に至るおそれがある。 | 神経外モノアミン濃度が高まると考えられる。 |
10.2 併用注意
| 尿のpHをアルカリ化する薬剤 炭酸水素ナトリウム等 | 本剤の作用が増強することがある。 | 本剤の活性体であるd-アンフェタミンの腎排泄が抑制され、半減期が延長する。 |
| 尿のpHを酸性化する薬剤 アスコルビン酸等 | 本剤の作用が減弱することがある。 | 本剤の活性体であるd-アンフェタミンの腎排泄が促進され、半減期が短縮する。 |
| セロトニン作用薬 選択的セロトニン再取り込み阻害剤(SSRI)、セロトニン・ノルアドレナリン再取り込み阻害剤(SNRI)、三環系抗うつ剤等 | まれにセロトニン症候群が起こることがある。 | 本剤のセロトニン再取り込み阻害作用及び神経終末からのセロトニン放出促進により、セロトニン作用が増強すると考えられる。 |
| メチルフェニデート塩酸塩 | メチルフェニデート塩酸塩を投与中の患者には本剤の投与を避けることが望ましい。本剤の作用が増強するおそれがある。 | 相加作用のおそれがある。 |
| イオフルパン(123I) | 本剤が線条体におけるイオフルパン(123I)の集積低下の原因となる可能性がある。画像を評価する際に留意すること。 | 本剤の活性体であるd-アンフェタミンがイオフルパン(123I)のドパミントランスポーターへの結合を低下させ、検査結果に干渉するおそれがある。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 ショック、アナフィラキシー(頻度不明)
ショック、アナフィラキシー(顔面蒼白、呼吸困難、そう痒等)があらわれることがある。
11.1.2 皮膚粘膜眼症候群(Stevens-Johnson症候群)(頻度不明)
11.1.3 心筋症(頻度不明)
11.1.4 依存性(頻度不明)
不適切な使用により精神的依存を生じることがあるので、観察を十分に行い、用量及び使用期間に注意し、慎重に投与すること。[8.12参照]
11.2 その他の副作用
| 5%以上 | 1〜5%未満 | 1%未満 | 頻度不明 | |
| 過敏症 | 発疹 | 過敏症、蕁麻疹、血管浮腫 | ||
| 循環器 | 頻脈 | 血圧上昇、動悸 | レイノー現象 | |
| 精神神経系 | 不眠(45.3%)、頭痛、めまい | 易刺激性、チック、眠気、感情不安定、激越 | 振戦、怒り、不安 | 多弁、リビドー減退、うつ病、不快気分、多幸症、歯ぎしり、自傷性皮膚症、精神病性障害、躁病、幻覚、攻撃性、落ち着きのなさ、精神運動亢進、痙攣、ジスキネジア、味覚異常 |
| 消化器 | 食欲減退(79.1%)、悪心、腹痛、下痢、嘔吐 | 便秘、口内乾燥 | 腹部不快感 | |
| その他 | 体重減少(25.6%) | 疲労感 | 霧視、散瞳、呼吸困難、好酸球性肝炎、多汗症、胸痛、びくびく感、発熱、勃起不全、鼻出血、脱毛症 |
13. 過量投与
13.1 症状
急性過量投与の症状は、落ち着きのなさ、振戦、反射亢進、頻呼吸、錯乱、攻撃性、幻覚、パニック状態、異常高熱、横紋筋融解等である。セロトニン症候群の発現も報告されている。通常、疲労及び抑うつは中枢神経系刺激後に生じる。心血管系への影響として不整脈、高血圧あるいは低血圧、循環虚脱等があらわれる。また、胃腸症状として悪心、嘔吐、下痢、腹部仙痛等があらわれる。致死的な中毒を起こす前には、通常、痙攣及び昏睡があらわれる。
13.2 処置
14. 適用上の注意
14.1 薬剤調製時の注意
PTP包装から取り出した無包装状態では、吸湿により品質に影響を及ぼすことが認められたため、分包しないこと。
14.2 薬剤交付時の注意
14.2.1 PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
14.2.2 本剤の投与にあたっては、本剤の目的以外への使用あるいは他人への譲渡をしないよう指導すること。[8.12参照]
14.2.3 本剤が不要となった場合には、医療機関又は薬局へ返却するなどの処置について適切に指導すること。
15. その他の注意
15.2 非臨床試験に基づく情報
本剤のがん原性試験は実施していない。活性体であるd-アンフェタミンのマウス及びラットのがん原性試験ではがん原性を示唆する所見は見られなかったが、これらのがん原性試験は臨床曝露量未満で実施されており、十分な安全域は担保されていない。
16. 薬物動態
16.1 血中濃度
16.1.1 健康成人
健康成人11例に本剤20mg注、50mg及び70mgを漸増法でそれぞれ1日1回空腹時5日間、計15日間反復経口投与したとき、各投与量における投与5日目の血漿中リスデキサンフェタミン及びd-アンフェタミン濃度推移を図16-1に、薬物動態パラメータを表16-1に示す。d-アンフェタミンは投与後3〜5時間でCmaxに達し、Cmax及びAUCは用量に比例して増加した。また、反復投与開始後5日以内に定常状態に達した1)。
図16-1 健康成人における平均リスデキサンフェタミン及びd-アンフェタミン血漿中濃度推移(反復投与:各投与量注における投与5日目)
| 測定成分 | 投与量(mg) | 例数 | Cmax※1(ng/mL) | AUC0-τ※1(ng・hr/mL) | Tmax※2(hr) |
| リスデキサンフェタミン | 20 | 11 | 8.82±2.44 | 10.50±2.69 | 1(1-1.5) |
| 50 | 33.58±10.19 | 41.32±10.52 | 1(1-2) | ||
| 70 | 10 | 47.27±19.94 | 65.89±23.09 | 1.5(1-3) | |
| d-アンフェタミン | 20 | 11 | 25.80±5.29 | 335.84±89.73 | 3(1.5-5) |
| 50 | 66.12±13.24 | 889.48±191.83 | 4(3-5) | ||
| 70 | 10 | 92.07±16.51 | 1280.56±290.06 | 5(3-8) |
16.1.2 小児患者
日本人及び外国人小児AD/HD患者(194例)から得られた血漿中d-アンフェタミン濃度データ(1365ポイント)を用いて母集団薬物動態解析を行った。その結果、見かけの全身クリアランスに対して体重及び民族が、見かけの分布容積に対して体重が統計学的に有意な共変量であった。また、日本人児童患者60例(6〜12歳)及び青少年患者19例(13〜17歳)に本剤30mg、50mg及び70mgを1日1回経口投与したとき、母集団薬物動態解析の結果に基づき推定した薬物動態パラメータは表16-2のとおりである2)。
| 投与群 | 児童(6〜12歳) | 青少年(13〜17歳) | ||||
| 例数 | Cmax(ng/mL) | AUC0-τ(ng・hr/mL) | 例数 | Cmax(ng/mL) | AUC0-τ(ng・hr/mL) | |
| 30mg | 16 | 66.7(50.4-99.6) | 1028(821.8-1487) | 5 | 47.7(33.0-54.4) | 750.1(518.3-883.9) |
| 50mg | 18 | 119(82.8-147) | 1885(1362-2278) | 5 | 77.3(59.5-89.6) | 1310(961.0-1500) |
| 70mg | 26 | 168(94.3-250) | 2669(1599-3711) | 9 | 118(102-129) | 1953(1563-2144) |
16.2 吸収
16.2.1 吸収に関与するトランスポーター
リスデキサンフェタミンは消化管から速やかに吸収され、その吸収には、ペプチドトランスポーターであるPEPT1が関与することが示唆されている3)(in vitro)。
16.2.2 食事の影響
健康成人18例に、本剤70mgを空腹時又は朝食後(高脂肪食)に単回経口投与した場合、d-アンフェタミンのTmaxは約1時間遅延したが、Cmax及びAUCに差は認められなかった4)(外国人データ)。
16.3 分布
16.3.1 蛋白結合率
d-アンフェタミンのヒト血漿蛋白結合率は約16%である5)。
16.4 代謝
16.4.1 リスデキサンフェタミンは、主に血中で活性体であるd-アンフェタミンに加水分解される6)。d-アンフェタミンは主に脱アミノ反応を経て馬尿酸や安息香酸に代謝され、また、一部4位水酸化反応でも代謝されることが報告されている。なお、4位水酸化反応にはCYP2D6が関与することが報告されている7)。
16.4.2 健康成人6例に14Cで標識したリスデキサンフェタミンメシル酸塩70mgを単回経口投与したとき、d-アンフェタミン、馬尿酸及び安息香酸(それぞれ投与量の41.5%、24.8%及び2.2%)が尿中代謝物として検出された8)(外国人データ)。
16.5 排泄
16.5.1 健康成人12例に本剤20mg注を単回経口投与したときのリスデキサンフェタミン及びd-アンフェタミンのT1/2の算術平均値(標準偏差)は、それぞれ0.44時間(0.01)及び9.65時間(1.48)であった1)。
16.5.2 健康成人6例に14Cで標識したリスデキサンフェタミンメシル酸塩70mgを単回経口投与したとき、投与後120時間までに投与放射能の96.4%が尿中に排泄され、糞中への排泄は0.3%未満であった。また、投与後48時間までに投与放射能の2.2%がリスデキサンフェタミンとして、41.5%がd-アンフェタミンとして、24.8%が馬尿酸として尿中に排泄された8)(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
軽度から高度の腎機能障害を有する被験者24例、血液透析を要する被験者8例及び腎機能正常者8例に本剤30mgを単回経口投与したときのd-アンフェタミン薬物動態パラメータを表16-3に示す。腎機能の低下に伴い、血漿中からの消失が遅延し、AUCが増大することが示された。また、リスデキサンフェタミン及びd-アンフェタミンは透析でほとんど除去されなかった9)(外国人データ)。[7.2、9.2.1参照]
| 投与群 | 例数 | Cmax※1(ng/mL) | AUC0-inf※1(ng・hr/mL) | Tmax※2(hr) | T1/2※1(hr) | |
| 腎機能正常者 | 8 | 32.2±5.3 | 597.9±44.5 | 3.5(3-4) | 12.1±2.5 | |
| 腎機能障害者 | 軽度 60≦eGFR<90 | 8 | 35.1±11.1 | 637.7±123.8 | 4(2-6) | 12.8±2 |
| 中等度 30≦eGFR<60 | 8 | 27.5±4.9 | 702.7±182.9 | 4(3-6) | 16.8±5.2 | |
| 高度 15≦eGFR<30 | 8 | 28.4±5.9 | 856.9±161.5 | 4(2-6) | 19.8±1.9 | |
| 血液透析※3 | 8 | 20.1±3.3 | 1126.3±437.9 | 4(2-8) | 38.2±16.5 | |
16.7 薬物相互作用
16.7.1 グアンファシン塩酸塩との併用
健康成人41例に、本剤50mgとグアンファシン塩酸塩徐放錠4mgの単回投与における薬物相互作用試験を実施したところ、本剤存在下でグアンファシンのCmaxは約19%増加したが、AUCに対する影響は認められなかった。また、グアンファシン塩酸塩徐放錠併用投与によるリスデキサンフェタミン及びd-アンフェタミンの薬物動態への影響は認められなかった10)(外国人データ)。
16.7.2 ベンラファキシン塩酸塩との併用
健康成人76例に、本剤70mgとCYP2D6基質であるベンラファキシン塩酸塩徐放性カプセル225mgの漸増反復投与における薬物相互作用試験を実施したところ、本剤存在下でベンラファキシンのCmaxは約10%、AUCは約13%増加した。また、ベンラファキシンの活性代謝物であるO-デスメチルベンラファキシンのCmaxは約9%、AUCは約17%減少した。ベンラファキシン塩酸塩徐放性カプセル併用投与によるリスデキサンフェタミン及びd-アンフェタミンの薬物動態への影響は認められなかった11)(外国人データ)。
注)本剤の承認用量は、通常1日30mg、症状により1日70mgを超えない範囲で適宜増減である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国内第II/III相試験(二重盲検試験)
小児AD/HD患者(6歳以上18歳未満)を対象に実施した第2/3相二重盲検並行群間比較試験において、主要評価項目であるm-ITT集団における投与4週時のADHD-RS-IV合計スコアのベースラインからの変化量は表17-1のとおりであり、本剤各群とプラセボ群との間に統計学的な有意差が認められた12)。[5.2、9.7参照]
| 投与群 | ベースライン | 投与4週後 | ベースラインからの変化量※2、3 | プラセボとの比較※3 | |||
| 例数 | 測定値※1 | 例数 | 測定値※1 | 群間差[95%信頼区間] | p値※4 | ||
| プラセボ | 19 | 37.95±7.40 | 19 | 34.68±10.73 | −2.78±2.25 | − | − |
| 30mg | 19 | 38.05±6.74 | 18 | 19.78±9.74 | −16.38±2.24 | −13.61[−19.80,−7.42] | <0.0001 |
| 50mg | 18 | 37.06±6.94 | 17 | 17.41±9.04 | −18.10±2.35 | −15.32[−21.65,−9.00] | <0.0001 |
| 70mg | 20 | 37.15±7.80 | 17 | 20.47±13.15 | −16.47±2.29 | −13.69[−19.98,−7.40] | <0.0001 |
副作用は、本剤30mg群で13/19例(68.4%)、本剤50mg群で18/18例(100.0%)、本剤70mg投与群で13/20例(65.0%)に認められた。主な副作用は、本剤30mg群では食欲減退8例(42.1%)、本剤50mg群では食欲減退14例(77.8%)、頭痛6例(33.3%)及び初期不眠症5例(27.8%)、本剤70mg群では食欲減退11例(55.0%)及び初期不眠症5例(25.0%)であった。
17.1.2 国内第III相試験(長期投与試験)
小児AD/HD患者(6歳以上18歳未満)を対象に実施した第3相長期投与試験において、有効性の評価尺度であるADHD-RS-IV合計スコアの推移(m-ITT集団)は表17-2のとおりであった13)。[5.2、9.7参照]
| 期間(週) | 継続例 | 新規例※3 | ||||
| プラセボ/本剤集団※1 | 本剤/本剤集団※2 | |||||
| 例数 | 合計スコア | 例数 | 合計スコア | 例数 | 合計スコア | |
| 0※4 | 19 | 34.84±10.76 | 50 | 29.78±10.88 | 63 | 31.35±8.04 |
| 1 | 19 | 29.79±8.32 | 50 | 23.10±10.57 | 63 | 20.37±10.05 |
| 2 | 19 | 26.63±9.41 | 50 | 18.86±10.09 | 62 | 18.10±9.94 |
| 3 | 18 | 25.22±10.14 | 50 | 16.78±10.38 | 61 | 15.11±10.36 |
| 5 | 17 | 23.76±10.21 | 48 | 15.92±10.12 | 61 | 12.67±9.06 |
| 9 | 16 | 23.38±9.54 | 49 | 14.96±9.91 | 58 | 12.93±9.28 |
| 13 | 16 | 22.13±8.96 | 47 | 15.28±10.06 | 58 | 11.72±9.11 |
| 17 | 15 | 20.67±8.80 | 46 | 15.15±10.40 | 57 | 10.82±8.41 |
| 29 | 15 | 18.80±10.01 | 43 | 13.77±9.97 | 55 | 10.40±9.17 |
| 41 | 15 | 16.47±8.16 | 40 | 14.75±9.89 | 51 | 8.67±8.00 |
| 53 | 13 | 15.23±7.72 | 40 | 14.80±10.78 | 51 | 8.49±7.37 |
| 最終評価時 | 19 | 18.63±9.31 | 50 | 15.00±10.13 | 63 | 10.40±8.66 |
副作用は全体で116/132例(87.9%)に認められた。主なものは、食欲減退97例(73.5%)、初期不眠症50例(37.9%)であった。
18. 薬効薬理
18.1 作用機序
リスデキサンフェタミンはプロドラッグであり、活性体であるd-アンフェタミンは、ノルアドレナリントランスポーター及びドパミントランスポーターに対する阻害作用、脳内シナプトソームからのノルアドレナリン及びドパミンの遊離作用、モノアミン酸化酵素Aに対する阻害作用を示し、前頭前皮質及び線条体における細胞外ノルアドレナリン及びドパミン濃度を増加させることによりシグナルを調節している可能性が示唆されているが、AD/HDの治療効果における詳細な作用機序は不明である14)。
18.2 薬理作用
幼若ラットにリスデキサンフェタミンを経口投与すると、衝動性行動の減少が認められた。AD/HDモデル動物である自然発症高血圧ラット及び6-hydroxydopamineにより脳内のドパミン神経を変性させたラットにd-アンフェタミンを腹腔内投与すると、多動性及び衝動性の改善が認められた。マウス及び自然発症高血圧ラットに低用量のd-アンフェタミンを腹腔内投与すると持続的注意の改善が認められた14)。
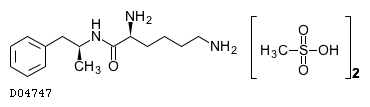
19. 有効成分に関する理化学的知見
19.1. リスデキサンフェタミンメシル酸塩
| 一般的名称 | リスデキサンフェタミンメシル酸塩 |
|---|---|
| 一般的名称(欧名) | Lisdexamfetamine Mesilate |
| 化学名 | (2S)-2,6-Diamino-N-[(2S)-1-phenylpropan-2-yl]hexanamide dimethanesulfonate |
| 分子式 | C15H25N3O・2CH4O3S |
| 分子量 | 455.59 |
| 融点 | 192〜198℃ |
| 物理化学的性状 | 白色〜淡黄白色の粉末又は塊である。 |
| 分配係数 | −1.76[1-オクタノール/水] |
| KEGG DRUG |
|
20. 取扱い上の注意
アルミピロー包装開封後は、湿気を避けて遮光して保存すること。
21. 承認条件
21.1 医薬品リスク管理計画を策定の上、適切に実施すること。
21.2 本剤が、注意欠陥/多動性障害(AD/HD)の診断、治療に精通した医師によって適切な患者に対してのみ処方されるとともに、薬物依存を含む本剤のリスク等について十分に管理できる医療機関及び薬局においてのみ取り扱われるよう、製造販売にあたって必要な措置を講じること。
21.3 使用実態下における乱用・依存性に関する評価が行われるまでの間は、他のAD/HD治療薬が効果不十分な場合にのみ使用されるよう必要な措置を講じること。
22. 包装
<ビバンセカプセル20mg>
30カプセル[10カプセル(PTP)×3]
<ビバンセカプセル30mg>
30カプセル[10カプセル(PTP)×3]
23. 主要文献
- 社内資料:健康成人の薬物動態試験(2019/3/26承認、申請資料概要2.7.6.2.1)
- 社内資料:小児AD/HD患者における母集団薬物動態解析(2019/3/26承認、申請資料概要2.7.2.3)
- 社内資料:非臨床薬物動態試験:吸収(2019/3/26承認、申請資料概要2.6.4.3)
- 社内資料:食事の影響試験(2019/3/26承認、申請資料概要2.7.6.1.1)
- Baggot,J.D.et al., Biochem.Pharmacol., 21, 1813-1816, (1972) »PubMed
- 社内資料:非臨床薬物動態試験:代謝(2019/3/26承認、申請資料概要2.6.4.5)
- Golub,M.et al., Birth Defects Res.B Dev.Reprod.Toxicol., 74, 471-584, (2005) »PubMed
- 社内資料:マスバランス試験(2019/3/26承認、申請資料概要2.7.6.2.2)
- 社内資料:腎機能障害者試験(2019/3/26承認、申請資料概要2.7.6.4.1)
- 社内資料:薬物相互作用試験−グアンファシン塩酸塩−(2019/3/26承認、申請資料概要2.7.6.5.2)
- 社内資料:薬物相互作用試験−ベンラファキシン塩酸塩−(2019/3/26承認、申請資料概要2.7.6.5.3)
- 社内資料:小児AD/HD患者の第2/3相二重盲検試験(2019/3/26承認、申請資料概要2.7.6.6.1)
- 社内資料:小児AD/HD患者の第3相長期投与試験(2019/3/26承認、申請資料概要2.7.6.6.13)
- 社内資料:効力を裏付ける試験(2019/3/26承認、申請資料概要2.6.2.2)
24. 文献請求先及び問い合わせ先
文献請求先
武田薬品工業株式会社
くすり相談室
〒103-8668
東京都中央区日本橋本町二丁目1番1号
電話:フリーダイヤル 0120-566-587 受付時間 9:00〜17:30(土日祝日・弊社休業日を除く)
製品情報問い合わせ先
武田薬品工業株式会社
くすり相談室
〒103-8668
東京都中央区日本橋本町二丁目1番1号
電話:フリーダイヤル 0120-566-587 受付時間 9:00〜17:30(土日祝日・弊社休業日を除く)
25. 保険給付上の注意
25.1 本製剤の使用に当たっての留意事項については、「リスデキサンフェタミンメシル酸塩製剤の使用に当たっての留意事項について」(平成31年3月26日付け薬生総発0326第1号・薬生薬審発0326第1号・薬生安発0326第8号・薬生監麻発0326第50号厚生労働省医薬・生活衛生局総務課長・医薬品審査管理課長・医薬安全対策課長・監視指導・麻薬対策課長通知)により通知されたところであるので、十分留意すること。(令和元年5月21日付け保医発0521第4号厚生労働省保険局医療課長通知)
25.2 本剤は厚生労働省告示第234号(令和2年6月1日付)に基づき、投薬量は1回30日分を限度とされている。
26. 製造販売業者等
26.1 製造販売元
武田薬品工業株式会社
〒540-8645
大阪市中央区道修町四丁目1番1号