医薬品情報
| 総称名 | ベネクレクスタ |
|---|---|
| 一般名 | ベネトクラクス |
| 欧文一般名 | Venetoclax |
| 製剤名 | ベネトクラクス錠 |
| 薬効分類名 | 抗悪性腫瘍剤 BCL-2阻害剤 |
| 薬効分類番号 | 4291 |
| ATCコード | L01XX52 |
| KEGG DRUG |
D10679
ベネトクラクス
|
| JAPIC | 添付文書(PDF) |
添付文書情報2025年11月 改訂(効能又は効果、用法及び用量変更)(第10版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ベネクレクスタ錠10mg | VENCLEXTA Tablets | アッヴィ | 4291062F1020 | 872.8円/錠 | 劇薬, 処方箋医薬品注) |
| ベネクレクスタ錠50mg | VENCLEXTA Tablets | アッヴィ | 4291062F2027 | 3956.6円/錠 | 劇薬, 処方箋医薬品注) |
| ベネクレクスタ錠100mg | VENCLEXTA Tablets | アッヴィ | 4291062F3023 | 7585.9円/錠 | 劇薬, 処方箋医薬品注) |
1. 警告
1.1 本剤は、緊急時に十分対応できる医療施設において、造血器悪性腫瘍の治療に対して十分な知識・経験を持つ医師のもとで、本剤の投与が適切と判断される症例についてのみ投与すること。また、治療開始に先立ち、患者又はその家族に有効性及び危険性を十分説明し、同意を得てから投与を開始すること。
2. 禁忌
次の患者には投与しないこと
4. 効能または効果
5. 効能または効果に関連する注意
<慢性リンパ性白血病(小リンパ球性リンパ腫を含む)>
<急性骨髄性白血病>
6. 用法及び用量
<未治療の慢性リンパ性白血病(小リンパ球性リンパ腫を含む)>
他の抗悪性腫瘍剤との併用において、通常、成人にはベネトクラクスとして、用量漸増期は第1週目に20mg、第2週目に50mg、第3週目に100mg、第4週目に200mg、第5週目に400mgをそれぞれ1日1回、7日間食後に経口投与する。その後の維持投与期は、400mgを1日1回、食後に経口投与する。なお、患者の状態により適宜減量する。
<再発又は難治性の慢性リンパ性白血病(小リンパ球性リンパ腫を含む)>
通常、成人にはベネトクラクスとして、用量漸増期は第1週目に20mg、第2週目に50mg、第3週目に100mg、第4週目に200mg、第5週目に400mgをそれぞれ1日1回、7日間食後に経口投与する。その後の維持投与期は、400mgを1日1回、食後に経口投与する。なお、患者の状態により適宜減量する。
<再発又は難治性のマントル細胞リンパ腫>
イブルチニブとの併用において、通常、成人にはベネトクラクスとして、用量漸増期は第1週目に20mg、第2週目に50mg、第3週目に100mg、第4週目に200mg、第5週目に400mgをそれぞれ1日1回、7日間食後に経口投与する。その後の維持投与期は、400mgを1日1回、食後に経口投与する。なお、患者の状態により適宜減量する。
<急性骨髄性白血病>
アザシチジン併用の場合
通常、成人にはベネトクラクスとして、用量漸増期は1日目に100mg、2日目に200mg、3日目に400mgをそれぞれ1日1回、食後に経口投与する。その後の維持投与期は、400mgを1日1回、食後に経口投与する。なお、患者の状態により適宜減量する。
シタラビン少量療法併用の場合
通常、成人にはベネトクラクスとして、用量漸増期は1日目に100mg、2日目に200mg、3日目に400mg、4日目に600mgをそれぞれ1日1回、食後に経口投与する。その後の維持投与期は、600mgを1日1回、食後に経口投与する。なお、患者の状態により適宜減量する。
7. 用法及び用量に関連する注意
<慢性リンパ性白血病(小リンパ球性リンパ腫を含む)>
7.1 本剤の投与により副作用が発現した場合には、以下の基準を参考に、本剤を休薬、減量、中止すること。なお、一定期間休薬後に再開する場合には、腫瘍崩壊症候群のリスク評価を行い、本剤の投与量を決定すること。[1.2、8.1、8.2、11.1.1、11.1.2参照]
| 副作用* | 処置 |
| Grade 4の血液毒性(好中球減少、血小板減少及びリンパ球減少を除く) | Grade 1以下又はベースラインに回復するまで休薬し、回復後は休薬前と同じ用量レベルで投与を再開する。 再開した後に再び発現した場合、Grade 1以下に回復するまで休薬し、回復後は休薬前より1段階低い用量レベルで投与を再開する。 |
| Grade 3又は4の好中球減少 | Grade 1以下又はベースラインに回復するまで休薬し、回復後は休薬前と同じ用量レベルで投与を再開する。感染を伴う場合、感染が消失した後に再開する。 再開した後に再び発現した場合、Grade 1以下に回復するまで休薬し、回復後は休薬前より1段階低い用量レベルで投与を再開する。 |
| Grade 3又は4の血小板減少 | Grade 1以下又はベースラインに回復するまで休薬し、回復後は休薬前と同じ用量レベルで投与を再開する。 再開した後に再び発現した場合、Grade 1以下に回復するまで休薬し、回復後は休薬前より1段階低い用量レベルで投与を再開する。 |
| 腫瘍崩壊症候群 | 腫瘍崩壊症候群が消失するまで休薬し、消失後は休薬前と同じ用量レベル又は1段階低い用量レベルで投与を再開する。 2週間以上の休薬を要した場合、休薬前より1段階低い用量レベルで投与を再開する。 |
| Grade 3又は4の非血液毒性(腫瘍崩壊症候群を除く) | Grade 1以下又はベースラインに回復するまで休薬し、回復後は休薬前と同じ用量レベルで投与を再開する。 再開した後に再び発現した場合、Grade 1以下又はベースラインに回復するまで休薬し、回復後は休薬前より1段階低い用量レベルで投与を再開する。 |
| 用量レベル | 本剤の1日用量 |
| 用量レベル 5 | 400mg |
| 用量レベル 4 | 300mg |
| 用量レベル 3 | 200mg |
| 用量レベル 2 | 100mg |
| 用量レベル 1 | 50mg |
| 用量レベル 0 | 20mg |
| 用量レベル −1 | 10mg |
<未治療の慢性リンパ性白血病(小リンパ球性リンパ腫を含む)>
7.4 オビヌツズマブ(遺伝子組換え)と併用する場合、通常、成人にはオビヌツズマブ(遺伝子組換え)を1サイクル目の1日目に100mg、2日目に900mg、8日目及び15日目に1000mg、2サイクル目以降は1日目に1000mgを点滴静注する。28日間を1サイクルとし、最大で6サイクル投与を繰り返す。なお、1サイクル目の22日目から、本剤の投与を開始すること。
7.5 イブルチニブと併用する場合、28日間を1サイクルとし、イブルチニブを3サイクル投与した後に、本剤の投与を開始すること。
7.6 本剤を12サイクルを超えて投与した場合の有効性及び安全性は確立していない。
<再発又は難治性の慢性リンパ性白血病(小リンパ球性リンパ腫を含む)>
7.7 リツキシマブ(遺伝子組換え)の投与が困難な場合を除き、維持投与期の開始からリツキシマブ(遺伝子組換え)と併用投与すること。
7.8 リツキシマブ(遺伝子組換え)以外の抗悪性腫瘍剤との併用による有効性及び安全性は確立していない。
<再発又は難治性のマントル細胞リンパ腫>
7.9 イブルチニブに対して本剤を24カ月を超えて上乗せ投与した場合の有効性及び安全性に関する情報は限られているため、「17.臨床成績」の項の内容を熟知した上で、ベネフィット・リスクを考慮して、本剤の投与継続の可否を慎重に検討すること。[17.1.6、17.1.7参照]
7.10 本剤の投与により副作用が発現した場合には、以下の基準を参考に、本剤を休薬、減量、中止すること。なお、一定期間休薬後に再開する場合には、腫瘍崩壊症候群のリスク評価を行い、本剤の投与量を決定すること。[1.2、8.1、8.3、11.1.1、11.1.2参照]
| 副作用* | 処置 |
| Grade 4の血液毒性(好中球減少及びリンパ球減少を除く) | Grade 1以下又はベースラインに回復するまで休薬し、回復後は休薬前と同じ用量レベルで投与を再開する。 再開した後に再び発現した場合、Grade 1以下に回復するまで休薬し、回復後は休薬前より1段階低い用量レベルで投与を再開する。 |
| Grade 3又は4の好中球減少 | Grade 1以下又はベースラインに回復するまで休薬し、回復後は休薬前と同じ用量レベルで投与を再開する。感染を伴う場合、感染が消失した後に再開する。 再開した後に再び発現した場合、Grade 1以下に回復するまで休薬し、回復後は休薬前より1段階低い用量レベルで投与を再開する。 |
| 腫瘍崩壊症候群 | 腫瘍崩壊症候群が消失するまで休薬し、消失後は休薬前と同じ用量レベル又は1段階低い用量レベルで投与を再開する。 48時間以上の休薬を要した場合、休薬前より1段階低い用量レベルで投与を再開する。 |
| Grade 3又は4の非血液毒性(腫瘍崩壊症候群を除く) | Grade 1以下又はベースラインに回復するまで休薬し、回復後は休薬前と同じ用量レベルで投与を再開する。 再開した後に再び発現した場合、Grade 1以下又はベースラインに回復するまで休薬し、回復後は休薬前より1段階低い用量レベルで投与を再開する。 |
| 用量レベル | 本剤の1日用量 |
| 用量レベル 5 | 400mg |
| 用量レベル 4 | 300mg |
| 用量レベル 3 | 200mg |
| 用量レベル 2 | 100mg |
| 用量レベル 1 | 50mg |
| 用量レベル 0 | 20mg |
| 用量レベル −1 | 10mg |
<急性骨髄性白血病>
7.13 本剤の投与により副作用が発現した場合には、以下の基準を参考に、本剤を休薬、中止すること。[8.1、11.1.2参照]
| 副作用* | 処置 |
| Grade 4の好中球減少 | 寛解達成後初回発現時:Grade 3以下に回復するまで休薬し、回復後は休薬前と同じ用量で投与を再開する。 寛解達成後2回目以降の発現時:Grade 3以下に回復するまで休薬し、回復後は休薬前と同じ用量で投与を再開するが、21日間投与した後、7日間休薬すること。 |
| Grade 4の血小板減少 | 寛解達成後初回発現時:Grade 2以下に回復するまで休薬し、回復後は休薬前と同じ用量で投与を再開する。 寛解達成後2回目以降の発現時:Grade 2以下に回復するまで休薬し、回復後は休薬前と同じ用量で投与を再開するが、21日間投与した後、7日間休薬すること。 |
8. 重要な基本的注意
<効能共通>
<慢性リンパ性白血病(小リンパ球性リンパ腫を含む)>
8.2 腫瘍崩壊症候群があらわれることがあるため、以下の点に注意すること。[1.2、7.1、11.1.1参照]
・本剤投与開始前に血液検査(カリウム、カルシウム、リン、尿酸、クレアチニン)を行い、電解質異常のある場合は本剤投与開始に先立ち補正を行うこと。
・本剤投与開始前から、高尿酸血症治療剤の投与を行うこと。
・本剤投与開始前に、X線(CT検査)等による腫瘍量の評価により、腫瘍崩壊症候群のリスク評価を行い、本剤投与開始前及び用量漸増期には、腫瘍量に応じて、以下の表1及び表2を参考に対応すること。なお、具体的な方法、検査頻度等は患者の状態を考慮して判断すること。
| 水分補給注1) | 本剤による治療開始の2日前から開始し、用量漸増期を通じて1.5〜2L/日を摂取する。 | |
| 血液検査頻度 | 20mg及び50mgの各初回投与時注2) | 投与前、投与6〜8時間後、投与24時間後 |
| その後の各漸増用量の初回投与時 | 投与前 | |
| 水分補給注1) | 本剤による治療開始の2日前から開始し、用量漸増期を通じて1.5〜2L/日摂取に加え、補液投与(可能であれば150〜200mL/時)を行う。 | |
| 血液検査頻度 | 20mg及び50mgの各初回投与時注2) | 投与前及び投与4、8、12、24時間後 |
| その後の各漸増用量の初回投与時 | 投与前、投与6〜8時間後、投与24時間後 | |
注1):経口摂取困難な場合は補液投与を行うこと。
注2):クレアチニンクリアランスが80mL/min未満の中腫瘍量の患者では、20mg及び50mgの各初回投与時には高腫瘍量の場合の表を参照すること。
・本剤投与開始後、2週間以上休薬した後に再開する場合には、本剤投与開始前及び用量漸増期と同様の腫瘍崩壊症候群のリスク評価及び予防措置を行うこと。
・維持投与期においては、定期的に血液検査(カリウム、カルシウム、リン、尿酸、クレアチニン)を行うこと。
<再発又は難治性のマントル細胞リンパ腫>
8.3 腫瘍崩壊症候群があらわれることがあるため、以下の点に注意すること。[1.2、7.10、11.1.1参照]
・本剤投与開始前に血液検査(カリウム、カルシウム、リン、尿酸、クレアチニン)を行い、電解質異常のある場合は本剤投与開始に先立ち補正を行うこと。
・本剤投与開始前から、高尿酸血症治療剤の投与を行うこと。
・本剤投与開始前に、X線(CT検査)等による腫瘍量の評価により、腫瘍崩壊症候群のリスク評価を行い、本剤投与開始前及び用量漸増期には、腫瘍量に応じて、以下の表3及び表4を参考に対応すること。なお、具体的な方法、検査頻度等は患者の状態を考慮して判断すること。
| 水分補給注1) | 本剤による治療開始の2日前から開始し、用量漸増期を通じて1.5〜2L/日を摂取する。 | |
| 血液検査頻度 | 20mg及び50mgの各初回投与時 | 投与前、投与6〜8時間後、投与24時間後 |
| その後の各漸増用量の初回投与時 | 投与前 | |
| 水分補給注1) | 本剤による治療開始の2日前から開始し、用量漸増期を通じて1.5〜2L/日摂取に加え、補液投与(可能であれば150〜200mL/時)を行う。 | |
| 血液検査頻度 | 20mg及び50mgの各初回投与時 | 投与前及び投与4、8、12、24時間後 |
| その後の各漸増用量の初回投与時 | 投与前、投与6〜8時間後、投与24時間後 | |
注1):経口摂取困難な場合は補液投与を行うこと。
・本剤投与開始後、用量漸増期に1週間以上休薬した後又は維持投与期に2週間以上休薬した後に再開する場合には、本剤投与開始前及び用量漸増期と同様の腫瘍崩壊症候群のリスク評価及び予防措置を行うこと。
・維持投与期においては、定期的に血液検査(カリウム、カルシウム、リン、尿酸、クレアチニン)を行うこと。
<急性骨髄性白血病>
8.4 腫瘍崩壊症候群があらわれることがあるため、以下の点に注意すること。[1.2、11.1.1参照]
・白血球数が25×103/μL未満となるよう、本剤開始前に調整を行うこと。
・本剤投与開始前に血液検査(カリウム、カルシウム、リン、尿酸、クレアチニン)を行い、電解質異常のある場合は本剤投与開始に先立ち補正を行うこと。
・本剤投与開始前から、高尿酸血症治療剤の投与を行うこと。
・本剤投与開始前及び用量漸増期には、以下の表5を参考に対応すること。また、本剤投与開始前に、腫瘍崩壊症候群のリスク評価を行い、腫瘍崩壊症候群の危険因子を有する患者の場合、頻回な検査の実施や本剤を減量して開始するなど、追加の予防策を考慮すること。なお、具体的な方法、検査頻度等は患者の状態を考慮して判断すること。
| 水分補給注1) | 本剤による治療開始前から用量漸増期を通じて1.5〜2L/日を摂取する。 | |
| 血液検査頻度 | 用量漸増期 | 投与前、投与6〜8時間後 |
| 用量漸増期最終日 (アザシチジン併用の場合400mg到達時。シタラビン少量療法併用の場合600mg到達時) | 上記に加え、投与24時間後 | |
・維持投与期においては、定期的に血液検査(カリウム、カルシウム、リン、尿酸、クレアチニン)を行うこと。
9. 特定の背景を有する患者に関する注意
9.3 肝機能障害患者
9.3.1 重度の肝機能障害(Child-Pugh分類C)の患者
減量を考慮するとともに、患者の状態をより慎重に観察し、有害事象の発現に十分注意すること。血中濃度が上昇し、副作用が強くあらわれるおそれがある。[16.6.1参照]
9.4 生殖能を有する者
9.4.1 妊娠する可能性のある女性には、本剤投与中及び最終投与後30日間において避妊する必要性及び適切な避妊法について説明すること。[9.5参照]
9.4.2 生殖可能な年齢の男性に本剤を投与する場合には、性腺に対する影響を考慮すること。動物実験(イヌ)において、本剤1日1回400mg投与した時の臨床曝露量の約0.5倍の曝露に相当する用量で精原細胞を標的とした精巣毒性が認められており、回復性は確認されていない1)。
9.5 妊婦
9.6 授乳婦
授乳しないことが望ましい。動物実験(ラット)において乳汁中への移行が認められている3)。
9.7 小児等
小児等を対象とした臨床試験は実施していない。
10. 相互作用
相互作用序文
本剤は主にCYP3Aにより代謝される。また、本剤はP-糖タンパク(P-gp)の基質であり、P-gpを阻害する。[16.4参照]
薬物代謝酵素用語
CYP3A
薬物代謝酵素用語
P-糖タンパク(P-gp)
10.1 併用禁忌
10.2 併用注意
| <慢性リンパ性白血病(小リンパ球性リンパ腫を含む)の維持投与期、再発又は難治性のマントル細胞リンパ腫の維持投与期、急性骨髄性白血病> 強いCYP3A阻害剤 クラリスロマイシン イトラコナゾール ボリコナゾール ポサコナゾール 等 [7.2、7.11、7.14、10.1、16.7.2、16.7.7、16.7.8参照] | 本剤の副作用が増強されるおそれがあるので、本剤を減量するとともに、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | これらの薬剤等がCYP3Aを阻害することにより、本剤の血中濃度が上昇する可能性がある。 |
| 中程度のCYP3A阻害剤 エリスロマイシン ジルチアゼム フルコナゾール 等 [7.2、7.11、7.14、16.7.8参照] | 本剤の副作用が増強されるおそれがあるので、本剤を減量するとともに、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | これらの薬剤等がCYP3Aを阻害することにより、本剤の血中濃度が上昇する可能性がある。 |
| グレープフルーツ含有食品 | 本剤の副作用が増強されるおそれがあるので、摂取しないよう注意すること。 | これらの薬剤等がCYP3Aを阻害することにより、本剤の血中濃度が上昇する可能性がある。 |
| 強い又は中程度のCYP3A誘導剤 カルバマゼピン リファンピシン エファビレンツ 等 セイヨウオトギリソウ(St.John's Wort、セント・ジョーンズ・ワート)含有食品 [16.7.3、16.7.8参照] | 本剤の効果が減弱するおそれがあるので、CYP3A誘導作用のない又は弱い薬剤への代替を考慮すること。 | これらの薬剤等がCYP3Aを誘導することにより、本剤の血中濃度が低下する可能性がある。 |
| 生ワクチン又は弱毒生ワクチン | 接種した生ワクチンの原病に基づく症状が発現した場合には適切な処置を行うこと。 | ワクチン接種に対する応答が不明であり、また、生ワクチンによる二次感染が否定できない。 |
| ワルファリン [16.7.5参照] | ワルファリンの作用が増強されるおそれがあるので、プロトロンビン時間国際標準比(INR)値等の血液凝固能の変動に十分注意すること。 | 機序は不明であるが、ワルファリンの血中濃度が上昇する可能性がある。 |
| P-gp阻害剤 シクロスポリン タクロリムス リファンピシン 等 [16.7.3参照] | 本剤の副作用が増強されるおそれがあるので、本剤の減量を考慮するとともに、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | これらの薬剤がP-gpを阻害することにより、本剤の血中濃度が上昇する可能性がある。 |
| 治療域の狭いP-gpの基質となる薬剤 ジゴキシン エベロリムス シロリムス 等 [16.7.6参照] | これらの薬剤の副作用が増強されるおそれがあるので、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | 本剤がP-gpを阻害することにより、これらの薬剤の血中濃度が上昇する可能性がある。 |
| アジスロマイシン [16.7.4参照] | 本剤の効果が減弱するおそれがあるので、併用を避けることが望ましい。 | 機序は不明であるが、本剤の血中濃度が低下する可能性がある。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど、適切な処置を行うこと。
11.1.1 腫瘍崩壊症候群(2.1%)
11.1.2 骨髄抑制
11.1.3 感染症(26.7%)
肺炎(8.6%)、敗血症(4.4%)等があらわれることがある。
11.2 その他の副作用
| 10%以上 | 10%未満 | |
| 循環器 | − | 心房粗動 |
| 消化器 | 悪心(20.1%) 下痢(22.3%) | 便秘 口内炎 腹痛 嘔吐 |
| 一般・全身障害及び投与部位の状態 | − | 疲労 無力症 |
| 肝胆道系障害 | − | 血中ビリルビン増加 胆嚢炎/胆石症 |
| 代謝及び栄養障害 | − | 体重減少 低カリウム血症 低マグネシウム血症 食欲減退 |
| 筋骨格系及び結合組織障害 | − | 関節痛 |
| 神経系障害 | − | 浮動性めまい/失神 頭痛 |
| 腎及び尿路障害 | − | 血中クレアチニン増加 |
| 呼吸器、胸郭及び縦隔障害 | − | 呼吸困難 |
| 血管障害 | − | 出血 低血圧 |
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
15. その他の注意
15.1 臨床使用に基づく情報
海外臨床試験において、皮膚有棘細胞癌、扁平上皮癌、基底細胞癌等の二次性悪性腫瘍が発現したとの報告がある4)。
16. 薬物動態
16.1 血中濃度
<慢性リンパ性白血病(小リンパ球性リンパ腫を含む)>
16.1.1 単回投与
再発又は難治性の慢性リンパ性白血病(小リンパ球性リンパ腫を含む)患者に本剤20〜200mgを食後に単回経口投与したときの薬物動態パラメータを以下に示す5)(外国人データ)。
| 用量(mg) | n | Tmaxa(h) | Cmax(μg/mL) | AUC∞(μg・h/mL) | t1/2b(h) |
| 20注) | 3 | 6.0(6.0-6.0) | 0.07±0.02 | 1.9,2.1c | 16.1,17.7c |
| 50注) | 50 | 6.0(2.0-18.2) | 0.26±0.12 | 5.2±3.0d | 19.0±6.4d |
| 100注) | 1 | 8.0 | 1.19 | 35.8 | 22.5 |
| 200注) | 2 | 6.0,8.0 | 0.73,1.57 | 23.1,76.0 | 30.9,50.9 |
16.1.2 反復投与
日本人の再発又は難治性の慢性リンパ性白血病(小リンパ球性リンパ腫を含む)患者に本剤を、用量漸増期は第1週目に20mg、第2週目に50mg、第3週目に100mg、第4週目に200mg、第5週目に400mgをそれぞれ1日1回、7日間食後に経口投与し、その後の維持投与期に400mgを1日1回、食後に経口投与したときのベネトクラクスの平均血漿中濃度推移及び薬物動態パラメータを以下に示す6)。
| 時間 | 用量(mg) | n | Tmaxa(h) | Cmax(μg/mL) | AUC24(μg・h/mL) |
| 第7週 1日目 | 400 | 6 | 7.0(6.0-8.0) | 2.67±1.20 | 39.0±17.4 |
| 第10週 1日目b | 400 | 6 | 5.0(4.0-8.0) | 1.49±0.32 | 23.0±8.53 |
図1:再発又は難治性の慢性リンパ性白血病(小リンパ球性リンパ腫を含む)患者に本剤を反復経口投与したときの血漿中濃度推移(平均値+標準偏差)
16.1.3 反復投与(イブルチニブ併用時)
未治療の慢性リンパ性白血病(小リンパ球性リンパ腫を含む)患者に、イブルチニブ420mgとの併用下で本剤を、第1週目に20mg、第2週目に50mg、第3週目に100mg、第4週目に200mg、第5週目に400mgをそれぞれ1日1回、7日間食後に経口投与し、その後400mgを1日1回、食後に経口投与したときの9週1日目のベネトクラクスの薬物動態パラメータを以下に示す7)(外国人データ)。
| 時間 | 用量(mg) | n | Tmaxa(h) | Cmax(μg/mL) | AUC24(μg・h/mL) |
| 第9週 1日目 | 400 | 131 | 6.0(0.0-8.1) | 3.53±1.99 | 59.0±39.0 |
<再発又は難治性のマントル細胞リンパ腫>
16.1.4 反復投与(イブルチニブ併用時)
日本人の再発又は難治性のマントル細胞リンパ腫患者に、イブルチニブ560mgとの併用下で本剤を、用量漸増期は第1週目に20mg、第2週目に50mg、第3週目に100mg、第4週目に200mg、第5週目に400mgをそれぞれ1日1回、7日間食後に経口投与し、その後の維持投与期に400mgを1日1回、食後に経口投与したときの第6週1日目のベネトクラクスの薬物動態パラメータを以下に示す8)。
| 時間 | 用量(mg) | n | Tmaxa(h) | Cmax(μg/mL) | AUC24(μg・h/mL) |
| 第6週 1日目 | 400 | 11 | 8.0(6.0-8.0) | 5.87±2.68 | 94.4±44.1 |
<急性骨髄性白血病>
16.1.5 反復投与
強力な寛解導入療法の適応とならない未治療の急性骨髄性白血病患者に、シタラビン少量療法を1〜10日目に投与するとともに本剤を、用量漸増期は2日目に50mg、3日目に100mg、4日目に200mg、5日目に400mg、6日目に600mgをそれぞれ1日1回、食後に経口投与し、その後の維持投与期に600mgを1日1回、食後に経口投与したときのベネトクラクスの平均血漿中濃度推移及び薬物動態パラメータを以下に示す9)(外国人データ)。
| 時間 | 用量(mg) | n | Tmaxa(h) | Cmax(μg/mL) | AUC24(μg・h/mL) |
| 10日目b | 600 | 7 | 4.0(4.0-6.0) | 2.04±1.45 | 33.3±27.5 |
| 18日目 | 600 | 7 | 7.0(3.5-8.0) | 2.92±2.15 | 51.8±36.9c |
図2:急性骨髄性白血病患者に本剤を反復経口投与したときの血漿中濃度推移(平均値+標準偏差)
16.2 吸収
16.2.1 食事の影響
健康被験者24例に本剤100mg注)を低脂肪食摂取後に単回経口投与したとき、空腹時投与と比較してベネトクラクスのCmax及びAUC∞はいずれも3.4倍に増加した。また、高脂肪食摂取後に単回経口投与したとき、空腹時投与と比較してベネトクラクスのCmaxは5.3倍、AUC∞は5.1倍に増加した10)(外国人データ)。
16.3 分布
ベネトクラクスのヒト血漿タンパク非結合型分率は0.01未満であり、ヒト血液/血漿中濃度比は0.57であった3)(in vitro)。
16.4 代謝
16.5 排泄
健康被験者4例に14C-ベネトクラクス200mg注)を食後に単回経口投与したとき、投与後9日までに投与量の99.9%超が糞中に回収され、尿中排泄は0.1%未満であった。糞中において未変化のベネトクラクスが占める割合は20.8%であった12)(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 肝機能障害患者
16.7 薬物相互作用
16.7.1 ケトコナゾール
非ホジキンリンパ腫患者11例にケトコナゾール(強いCYP3A阻害剤、経口剤:国内未承認)400mgを1日1回7日間投与時に本剤50mg注)を食後に併用投与したとき、ベネトクラクスのCmaxは2.3倍、AUC∞は6.4倍に増加した14)(外国人データ)。
16.7.2 リトナビル
16.7.3 リファンピシン
単回投与
健康被験者11例にリファンピシン(P-gp阻害剤)600mgを単回投与時に本剤200mg注)を食後に併用投与したとき、ベネトクラクスのCmaxは2.1倍、AUC∞は1.8倍に増加した16)(外国人データ)。
反復投与
16.7.4 アジスロマイシン
16.7.5 ワルファリン
16.7.6 ジゴキシン
16.7.7 ポサコナゾール
16.7.8 イトラコナゾール、エリスロマイシン、フルコナゾール、エファビレンツ(生理学的薬物動態モデルによるシミュレーション)
16.7.9 その他
注)本剤の承認用法・用量は、慢性リンパ性白血病(小リンパ球性リンパ腫を含む)に対し「用量漸増期は第1週目に20mg、第2週目に50mg、第3週目に100mg、第4週目に200mg、第5週目に400mgをそれぞれ1日1回7日間投与、その後の維持投与期は、400mgを1日1回投与する。」又は急性骨髄性白血病に対しアザシチジンとの併用の場合は「用量漸増期は1日目に100mg、2日目に200mg、3日目に400mgをそれぞれ1日1回投与、その後の維持投与期は、400mgを1日1回投与する。」又はシタラビン少量療法併用の場合は「用量漸増期は1日目に100mg、2日目に200mg、3日目に400mg、4日目に600mgをそれぞれ1日1回投与、その後の維持投与期は、600mgを1日1回投与する。」である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
<慢性リンパ性白血病(小リンパ球性リンパ腫を含む)>
17.1.1 海外第III相試験(MURANO試験)
1レジメン以上の前治療歴を有する再発又は難治性の慢性リンパ性白血病患者389例を対象とし、本剤及びリツキシマブの併用療法(V+R)をベンダムスチン及びリツキシマブの併用療法(BR)と比較するランダム化非盲検第III相試験である。V+R群では本剤の用量漸増期注1)完了後、本剤を1日1回400mgで病態の悪化等が認められるまで最大2年間継続投与した。リツキシマブ注2)は28日を1サイクルとし、最大投与回数は6サイクルとして投与した。BR群ではベンダムスチンを1回量70mg/m2で2日間投与した。28日を1サイクルとし、最大投与回数は6サイクルとした。主要評価項目である治験責任医師判定の無増悪生存期間において、V+R群はBR群に対して統計学的に有意な延長を認めた(データカットオフ日:2017年5月8日)。
注1)本剤の漸増方法(用量漸増期):第1週目に20mg、第2週目に50mg、第3週目に100mg、第4週目に200mg、第5週目に400mgをそれぞれ1日1回、7日間食後に経口投与する。
注2)リツキシマブは初回に1回量375mg/m2、2回目以降は1回量500mg/m2を28日ごとに投与した。V+R群では本剤の用量漸増期完了後にリツキシマブの投与を開始し、BR群ではベンダムスチンの投与開始時にリツキシマブの投与を開始した。
| V+R群 | BR群 | |
| 症例数 | 194 | 195 |
| イベント発現例数 | 32 | 114 |
| 無増悪生存期間中央値(月) (95%信頼区間) | 未到達 | 17.0 (15.5-21.6) |
| ハザード比a (95%信頼区間) | 0.17(0.11-0.25) | |
| p値b | p<0.0001 | |
図1:無増悪生存期間のKaplan-Meier曲線
17.1.2 国内第I/II相試験(M13-834試験 Arm D)
再発又は難治性の慢性リンパ性白血病患者6例を対象として本剤とリツキシマブを併用投与した単群非盲検第II相試験である。本剤は20mgより投与を開始し400mg1日1回まで漸増を行った後、400mg1日1回投与を病態の悪化等が認められるまで継続した。リツキシマブは本剤の漸増期間完了後に投与を開始し、初回に1回量375mg/m2、2回目以降は1回量500mg/m2を28日ごとに投与した。28日を1サイクルとし、最大投与回数は6サイクルとした。主要評価項目である奏効率は66.7%(4/6例)(95%CI:22.3-95.7%)であった。
本剤を投与された6例のうち、6例(100%)にベネトクラクスとの因果関係のある副作用が認められた。主な副作用は、好中球減少症4例(66.7%)、悪心4例(66.7%)、白血球減少症3例(50.0%)、リンパ球減少症3例(50.0%)等であった6)。
本剤を投与された6例のうち、6例(100%)にベネトクラクスとの因果関係のある副作用が認められた。主な副作用は、好中球減少症4例(66.7%)、悪心4例(66.7%)、白血球減少症3例(50.0%)、リンパ球減少症3例(50.0%)等であった6)。
17.1.3 海外第III相試験(BO25323[CLL14]試験)無作為化パート
未治療の慢性リンパ性白血病患者注3)432例を対象とし、本剤及びオビヌツズマブ併用療法(V+O)をオビヌツズマブ及びchlorambucil(国内未承認)の併用療法(OC)と比較するランダム化非盲検第III相試験である。V+O群では、1サイクルを28日間として、オビヌツズマブ1000mgをサイクル1の1日目、8日目及び15日目、並びにその後の各サイクルの1日目に投与し、計6サイクル投与した(初回投与は1日目及び2日目に100mg及び900mgに分割することを可能とした)。本剤は、サイクル1の22日目から投与を開始し、用量漸増期注1)完了後、1日1回400mgで病態の悪化等が認められるまで最大12サイクル継続投与した。主要評価項目である治験責任医師判定による無増悪生存期間において、V+O群はOC群に対して統計学的に有意な延長を認めた。(データカットオフ日:2018年8月17日)
注3)併存疾患を有する([1]クレアチニンクリアランスが70mL/min未満、[2]Cumulative Illness Rating Scale(CIRS)スコアが6超の少なくとも1つを満たす)患者が対象とされた。
| V+O群 | OC群 | |
| 症例数 | 216 | 216 |
| イベント発現例数 | 30 | 77 |
| 無増悪生存期間中央値(月) (95%信頼区間) | 未到達 | 未到達 (31.1-未到達) |
| ハザード比a (95%信頼区間) | 0.35(0.23-0.53) | |
| p値b | p<0.0001 | |
図2:無増悪生存期間のKaplan-Meier曲線
17.1.4 海外第III相試験(CLL3011[GLOW]試験)
未治療の慢性リンパ性白血病患者注4)211例を対象とし、本剤及びイブルチニブの併用療法(V+I)をオビヌツズマブ及びchlorambucil(国内未承認)併用療法(OC)と比較するランダム化非盲検第III相試験である。V+I群では、1サイクルを28日間として、イブルチニブを420mg1日1回で3サイクルの単剤投与から開始し、15サイクルまで投与した。本剤は、サイクル4の1日目から投与を開始し、用量漸増期注1)完了後、1日1回400mgで病態の悪化等が認められるまで最大12サイクル継続投与した(サイクル15まで)。主要評価項目である独立評価委員会(IRC)判定による無増悪生存期間において、V+I群はOC群に対して統計学的に有意な延長を認めた。(データカットオフ日:2021年2月26日)
注4)65歳以上、又は65歳未満で併存疾患を有する([1]クレアチニンクリアランスが70mL/min未満、[2]Cumulative Illness Rating Scale(CIRS)スコアが6超の少なくとも1つを満たす)患者が対象とされた。
| V+I群 | OC群 | |
| 症例数 | 106 | 105 |
| イベント発現例数 | 22 | 67 |
| 無増悪生存期間中央値(月) (95%信頼区間) | 未到達 | 21.0 (16.6-24.7) |
| ハザード比a (95%信頼区間) | 0.22(0.13-0.36) | |
| p値b | p<0.0001 | |
図3:無増悪生存期間のKaplan-Meier曲線
17.1.5 国内第II相試験(M20-353試験)
未治療の慢性リンパ性白血病患者注4)10例(各コホート)を対象として本剤とオビヌツズマブを併用投与(V+O)(コホート1)又はイブルチニブを併用投与(V+I)(コホート2)した非対照非盲検第II相試験である。
コホート1では、1サイクルを28日間として、オビヌツズマブ1000mgをサイクル1の1日目、8日目及び15日目、並びにその後の各サイクルの1日目に投与し、計6サイクル投与した(初回投与は1日目及び2日目に100mg及び900mgに分割することを可能とした)。本剤は、サイクル1の22日目から投与を開始し、用量漸増期注1)完了後、1日1回400mgで病態の悪化等が認められるまで最大12サイクル継続投与した。
コホート2では、1サイクルを28日間として、イブルチニブを420mg1日1回で3サイクルの単剤投与から開始し、15サイクルまで投与した。本剤は、サイクル4の1日目から投与を開始し、用量漸増期注1)完了後、1日1回400mgで病態の悪化等が認められるまで最大12サイクル継続投与した(サイクル15まで)。
主要評価項目である独立評価委員会(IRC)判定による完全奏効(CR)及び骨髄回復が不完全な完全奏効(CRi)率は、コホート1(V+O)では90.0%(9/10例)(95%CI:55.5-99.7%)、コホート2(V+I)では60.0%(6/10例)(95%CI:26.2-87.8%)であった。
コホート1(V+O)10例のうち、9例(90.0%)にベネトクラクスとの因果関係のある副作用が認められた。主な副作用は、好中球数減少4例(40.0%)、悪心4例(40.0%)、貧血2例(20.0%)、好中球減少症2例(20.0%)、下痢2例(20.0%)、倦怠感2例(20.0%)、血小板数減少2例(20.0%)及び白血球数減少2例(20.0%)等であった。コホート2(V+I)10例のうち、9例(90.0%)にベネトクラクスとの因果関係のある副作用が認められた。主な副作用は、好中球数減少4例(40.0%)、悪心4例(40.0%)、好中球減少症3例(30.0%)、下痢3例(30.0%)、白血球数減少3例(30.0%)及び発疹3例(30.0%)等であった26)。[5.1、7.3参照]
コホート1では、1サイクルを28日間として、オビヌツズマブ1000mgをサイクル1の1日目、8日目及び15日目、並びにその後の各サイクルの1日目に投与し、計6サイクル投与した(初回投与は1日目及び2日目に100mg及び900mgに分割することを可能とした)。本剤は、サイクル1の22日目から投与を開始し、用量漸増期注1)完了後、1日1回400mgで病態の悪化等が認められるまで最大12サイクル継続投与した。
コホート2では、1サイクルを28日間として、イブルチニブを420mg1日1回で3サイクルの単剤投与から開始し、15サイクルまで投与した。本剤は、サイクル4の1日目から投与を開始し、用量漸増期注1)完了後、1日1回400mgで病態の悪化等が認められるまで最大12サイクル継続投与した(サイクル15まで)。
主要評価項目である独立評価委員会(IRC)判定による完全奏効(CR)及び骨髄回復が不完全な完全奏効(CRi)率は、コホート1(V+O)では90.0%(9/10例)(95%CI:55.5-99.7%)、コホート2(V+I)では60.0%(6/10例)(95%CI:26.2-87.8%)であった。
コホート1(V+O)10例のうち、9例(90.0%)にベネトクラクスとの因果関係のある副作用が認められた。主な副作用は、好中球数減少4例(40.0%)、悪心4例(40.0%)、貧血2例(20.0%)、好中球減少症2例(20.0%)、下痢2例(20.0%)、倦怠感2例(20.0%)、血小板数減少2例(20.0%)及び白血球数減少2例(20.0%)等であった。コホート2(V+I)10例のうち、9例(90.0%)にベネトクラクスとの因果関係のある副作用が認められた。主な副作用は、好中球数減少4例(40.0%)、悪心4例(40.0%)、好中球減少症3例(30.0%)、下痢3例(30.0%)、白血球数減少3例(30.0%)及び発疹3例(30.0%)等であった26)。[5.1、7.3参照]
<再発又は難治性のマントル細胞リンパ腫>
17.1.6 海外第III相試験(SYMPATICO[PCYC-1143-CA]試験)無作為化パート
再発又は難治性のマントル細胞リンパ腫患者267例を対象とし、本剤及びイブルチニブの併用療法(V+I)をプラセボ及びイブルチニブの併用療法(P+I)と比較するランダム化二重盲検第III相試験である。V+I群では本剤の用量漸増期注1)完了後、本剤を1日1回400mgで病態の悪化等が認められるまで最大2年間継続投与した。イブルチニブは用量漸増期から併用し、1日1回560mgで病態の悪化等が認められるまで継続投与した。主要評価項目である治験責任医師判定の無増悪生存期間(PFS)において、V+I群はP+I群に対して統計学的に有意な延長を認めた(データカットオフ日:2023年5月22日)。
注1)本剤の漸増方法(用量漸増期):第1週目に20mg、第2週目に50mg、第3週目に100mg、第4週目に200mg、第5週目に400mgをそれぞれ1日1回、7日間食後に経口投与する。
| V+I群 | P+I群 | |
| 症例数 | 134 | 133 |
| イベント発現例数 | 73 | 94 |
| 無増悪生存期間中央値(月) (95%信頼区間) | 31.9 (22.8-47.0) | 22.1 (16.5-29.5) |
| ハザード比a (95%信頼区間) | 0.645 (0.474-0.878) | |
| p値b | 0.0052 | |
図4:無増悪生存期間のKaplan-Meier曲線
17.1.7 国内第II相試験(M20-075試験)
再発又は難治性のマントル細胞リンパ腫患者13例を対象とし、本剤とイブルチニブを併用投与した単群非盲検第II相試験である。本剤の用量漸増期注1)完了後、本剤を1日1回400mgで病態の悪化等が認められるまで最大2年間継続投与した。イブルチニブは用量漸増期から併用し、1日1回560mgで病態の悪化等が認められるまで継続投与した。主要評価項目である独立評価委員会(IRC)による完全奏効(CR)率は83.3%(10/12例)(95%信頼区間:51.6-97.9%)であった(データカットオフ日:2022年2月9日)。
注1)本剤の漸増方法(用量漸増期):第1週目に20mg、第2週目に50mg、第3週目に100mg、第4週目に200mg、第5週目に400mgをそれぞれ1日1回、7日間食後に経口投与する。
<急性骨髄性白血病>
17.1.8 国際共同第III相試験(Viale-A[M15-656]試験)
強力な寛解導入療法の適応とならない未治療の急性骨髄性白血病患者433例(無作為割付例;日本人患者37例を含む)を対象とし、本剤及びアザシチジンの併用療法(V+AZA)をプラセボ及びアザシチジンの併用療法(P+AZA)と比較するランダム化二重盲検第III相試験である。第1、2及び3日目にそれぞれ本剤100、200及び400mg又はプラセボを1日1回食後に経口投与した後、本剤400mg又はプラセボを1日1回継続投与した。アザシチジンは28日を1サイクルとし、各サイクルの1〜7日目に75mg/m2を1日1回静脈内又は皮下投与した。主要評価項目の1つである全生存期間(OS)において、V+AZA群はP+AZA群に対して統計学的に有意な延長を認めた(データカットオフ日:2020年1月4日)。もう1つの主要評価項目である治験責任医師判定の複合的完全寛解(完全寛解[CR]+血球数回復が不完全な完全寛解[CRi])率において、V+AZA群はP+AZA群に対して統計学的に有意に高値であった(データカットオフ日:2018年10月1日)。
| V+AZA群 | P+AZA群 | |
| 全生存期間 | ||
| 症例数a | 286 | 145 |
| イベント発現例数 | 161 | 109 |
| 中央値(月) (95%信頼区間) | 14.7 (11.9-18.7) | 9.6 (7.4-12.7) |
| ハザード比b (95%信頼区間) | 0.662 (0.518-0.845) | |
| p値c | p<0.001 | |
| 複合的完全寛解率 | ||
| 症例数d | 147 | 79 |
| CR+CRi 例数(%) (95%信頼区間e) | 96(65.3) [57.0,73.0] | 20(25.3) [16.2,36.4] |
| p値f | p<0.001 | |
図5:全生存期間のKaplan-Meier曲線
17.1.9 国際共同第III相試験(Viale-C[M16-043]試験)
強力な寛解導入療法の適応とならない未治療の急性骨髄性白血病患者211例(日本人患者27例を含む)を対象とし、本剤及び低用量シタラビンの併用療法(V+LDAC)をプラセボ及び低用量シタラビンの併用療法(P+LDAC)と比較するランダム化二重盲検第III相試験である。第1、2、3及び4日目にそれぞれ本剤100、200、400及び600mg又はプラセボを1日1回経口投与した後、本剤600mg又はプラセボを1日1回継続投与した。低用量シタラビンは28日を1サイクルとし、各サイクルの1〜10日目に20mg/m2を1日1回皮下投与した。主要評価項目である全生存期間(OS)において、V+LDAC群はP+LDAC群に対して統計学的に有意な延長を示さなかった(データカットオフ日:2019年2月15日)。なお、治験責任医師判定の複合的完全寛解(完全寛解[CR]+血球数回復が不完全な完全寛解[CRi])率は、V+LDAC群では47.6%(68/143例)(95%信頼区間:39.1-56.1%)、P+LDAC群では13.2%(9/68例)(95%信頼区間:6.2-23.6%)であった。
| V+LDAC群 | P+LDAC群 | |
| 症例数 | 143 | 68 |
| イベント発現例数 | 86 | 47 |
| 全生存期間中央値(月) (95%信頼区間) | 7.2 (5.6-10.1) | 4.1 (3.1-8.8) |
| ハザード比a (95%信頼区間) | 0.749(0.524-1.071) | |
| p値b | p=0.114 | |
図6:全生存期間のKaplan-Meier曲線
18. 薬効薬理
18.1 作用機序
ベネトクラクスは抗アポトーシス作用を有するBcl-2に結合し、抗アポトーシス作用を阻害することによりアポトーシスを誘導すると考えられる33)。
18.2 抗腫瘍作用
ベネトクラクスは慢性リンパ性白血病(CLL)患者由来CLL細胞に対して増殖抑制作用を示した34)。
19. 有効成分に関する理化学的知見
19.1. ベネトクラクス
| 一般的名称 | ベネトクラクス |
|---|---|
| 一般的名称(欧名) | Venetoclax |
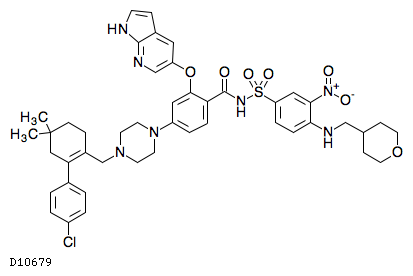
| 化学名 | 4-(4-{[2-(4-クロロフェニル)-4,4-ジメチルシクロヘキサ-1-エン-1-イル]メチル}ピペラジン-1-イル)-N-[(3-ニトロ-4-{[(オキサン-4-イル)メチル]アミノ}フェニル)スルホニル]-2-[(1H-ピロロ[2,3-b]ピリジン-5-イル)オキシ]ベンズアミド |
| 分子式 | C45H50ClN7O7S |
| 分子量 | 868.44 |
| 物理化学的性状 | 淡黄色〜黄色又は暗黄色の粉末である。 |
| KEGG DRUG |
|
21. 承認条件
<効能共通>
21.1 医薬品リスク管理計画を策定の上、適切に実施すること。
<急性骨髄性白血病>
21.2 国内での治験症例が極めて限られていることから、製造販売後、一定数の症例に係るデータが集積されるまでの間は、全症例を対象に使用成績調査を実施することにより、本剤使用患者の背景情報を把握するとともに、本剤の安全性及び有効性に関するデータを早期に収集し、本剤の適正使用に必要な措置を講じること。
22. 包装
<ベネクレクスタ錠10mg>
7錠[1錠(PTP)×7]
<ベネクレクスタ錠50mg>
7錠[1錠(PTP)×7]
<ベネクレクスタ錠100mg>
7錠[7錠(PTP)×1]
23. 主要文献
- 社内資料:反復投与毒性試験(2019年9月20日承認,CTD 2.6.6.3)
- 社内資料:生殖発生毒性試験(2019年9月20日承認,CTD 2.6.6.6)
- 社内資料:分布・排泄に関する検討(2019年9月20日承認,CTD 2.6.4.4)
- Seymour JF,et al., N Engl J Med., 378, 1107-1120, (2018) »PubMed »DOI
- 社内資料:海外第I相試験(M12-175試験)(2019年9月20日承認,CTD 2.7.2.2)
- 社内資料:国内第I/II相試験(M13-834試験)(2019年9月20日承認,CTD 2.7.2.2、2.7.3.1)
- 社内資料:海外第II相試験(1142試験)(2025年11月20日承認,CTD 2.7.2.2)
- 社内資料:国内第II相試験(M20-075試験)(2025年3月27日承認,CTD 2.7.2.2)
- 社内資料:海外第I/II相試験(M14-387試験)(2021年3月23日承認,CTD 2.7.2.2)
- 社内資料:食事の影響に関する試験(2019年9月20日承認,CTD 2.7.2.3)
- 社内資料:代謝に関する検討(2019年9月20日承認,CTD 2.6.4.5)
- 社内資料:マスバランス試験(2019年9月20日承認,CTD 2.7.2.2)
- 社内資料:肝機能障害患者における試験(2019年9月20日承認,CTD 2.7.2.2)
- 社内資料:ケトコナゾールとの薬物相互作用試験(2019年9月20日承認,CTD 2.7.2.2)
- 社内資料:リトナビルとの薬物相互作用試験(2019年9月20日承認,CTD 2.7.2.2)
- 社内資料:リファンピシンとの薬物相互作用試験(2019年9月20日承認,CTD 2.7.2.2)
- 社内資料:アジスロマイシンとの薬物相互作用試験(2019年9月20日承認,CTD 2.7.2.2)
- 社内資料:ワルファリンとの薬物相互作用試験(2019年9月20日承認,CTD 2.7.2.2)
- 社内資料:ジゴキシンとの薬物相互作用試験(2019年9月20日承認,CTD 2.7.2.2)
- 社内資料:海外第Ib相(M14-358)試験(2021年3月23日承認,CTD 2.7.2.2)
- 社内資料:生理学的薬物動態モデルによるシミュレーション(2019年9月20日承認,CTD 2.7.2.3)
- 社内資料:トランスポーターを介した薬物相互作用試験(2019年9月20日承認,CTD 2.6.4.7)
- 社内資料:海外第III相試験(MURANO試験)(2019年9月20日承認,CTD 2.7.3.1)
- 社内資料:海外第III相試験(BO25323試験)(2025年11月20日承認,CTD 2.7.3)
- 社内資料:海外第III相試験(CLL3011試験)(2025年11月20日承認,CTD 2.7.3)
- 社内資料:国内第II相試験(M20-353試験))(2025年11月20日承認,CTD 2.7.3)
- 社内資料:海外第III相試験(SYMPATICO試験)(2025年3月27日承認,CTD 2.7.3)
- 社内資料:国内第II相試験(M20-075試験)(2025年3月27日承認,CTD 2.7.3)
- DiNardo CD,et al., N Engl J Med., 383, 617-629, (2020) »PubMed
- 社内資料:海外第III相試験(VIALE-A[M15-656]試験)(2021年3月23日承認,CTD 2.7.3)
- Wei AH,et al., Blood., 135, 2137-2145, (2020) »PubMed
- 社内資料:海外第III相試験(VIALE-C[M16-043]試験)(2021年3月23日承認,CTD 2.7.3)
- 社内資料:in vitro薬理試験(2019年9月20日承認,CTD 2.6.2.2)
- 社内資料:ex vivo薬理試験(2019年9月20日承認,CTD 2.6.2.2.3)
24. 文献請求先及び問い合わせ先
文献請求先
アッヴィ合同会社
くすり相談室
〒108-0023
東京都港区芝浦3-1-21
電話:フリーダイヤル 0120-587-874
製品情報問い合わせ先
アッヴィ合同会社
くすり相談室
〒108-0023
東京都港区芝浦3-1-21
電話:フリーダイヤル 0120-587-874
26. 製造販売業者等
26.1 製造販売元
アッヴィ合同会社
東京都港区芝浦3-1-21