医薬品情報
| 総称名 | ユリス |
|---|---|
| 一般名 | ドチヌラド |
| 欧文一般名 | Dotinurad |
| 製剤名 | ドチヌラド |
| 薬効分類名 | 選択的尿酸再吸収阻害薬 高尿酸血症治療剤 |
| 薬効分類番号 | 3949 |
| ATCコード | M04AB06 |
| KEGG DRUG |
D11071
ドチヌラド
|
| JAPIC | 添付文書(PDF) |
添付文書情報2024年12月 改訂(第6版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ユリス錠0.5mg | URECE Tablets 0.5mg | 富士薬品 | 3949005F1022 | 22.9円/錠 | 処方箋医薬品 |
| ユリス錠1mg | URECE Tablets 1mg | 富士薬品 | 3949005F2029 | 41.7円/錠 | 処方箋医薬品 |
| ユリス錠2mg | URECE Tablets 2mg | 富士薬品 | 3949005F3025 | 74.2円/錠 | 処方箋医薬品 |
2. 禁忌
次の患者には投与しないこと
2.1 本剤の成分に対し過敏症の既往歴のある患者
4. 効能または効果
痛風、高尿酸血症
5. 効能または効果に関連する注意
6. 用法及び用量
通常、成人にはドチヌラドとして1日0.5mgより開始し、1日1回経口投与する。その後は血中尿酸値を確認しながら必要に応じて徐々に増量する。維持量は通常1日1回2mgで、患者の状態に応じて適宜増減するが、最大投与量は1日1回4mgとする。
7. 用法及び用量に関連する注意
8. 重要な基本的注意
8.1 本剤は尿酸降下薬であり、痛風関節炎(痛風発作)発現時に血中尿酸値を低下させると痛風関節炎(痛風発作)を増悪させるおそれがある。本剤投与前に痛風関節炎(痛風発作)が認められた場合は、症状がおさまるまで、本剤の投与を開始しないこと。
また、本剤投与中に痛風関節炎(痛風発作)が発現した場合には、本剤の用量を変更することなく投与を継続し、症状によりコルヒチン、非ステロイド性抗炎症剤、副腎皮質ステロイド等を併用すること。[7.参照]
また、本剤投与中に痛風関節炎(痛風発作)が発現した場合には、本剤の用量を変更することなく投与を継続し、症状によりコルヒチン、非ステロイド性抗炎症剤、副腎皮質ステロイド等を併用すること。[7.参照]
8.2 本剤の薬理作用により特に投与初期に尿酸排泄量が増大することから、尿が酸性の場合には、患者に尿路結石及びこれに由来する血尿、腎仙痛等の症状を起こす可能性があるので、これを防止するため、水分の摂取による尿量の増加及び尿のアルカリ化をはかること。なお、この場合には、患者の酸・塩基平衡に注意すること。
8.3 他の尿酸排泄促進薬において重篤な肝障害が報告されていることから、本剤投与中は、定期的に肝機能検査を行うなど、患者の状態を十分に観察すること。[9.3参照]
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 尿路結石を伴う患者
治療上やむを得ないと判断される場合を除き、投与しないこと。本剤の薬理作用から、尿中尿酸排泄量の増大により、尿路結石の症状を悪化させるおそれがある。
なお、臨床試験では、尿路結石を伴う患者への投与は行われていない。
なお、臨床試験では、尿路結石を伴う患者への投与は行われていない。
9.2 腎機能障害患者
9.2.1 重度の腎機能障害患者(eGFRが30mL/min/1.73m2未満)
他剤での治療を考慮すること。本剤は腎近位尿細管において作用するため、腎機能障害の程度に応じて、有効性が減弱する可能性がある。特に、乏尿又は無尿の患者においては、有効性が期待できないことから、本剤の投与は避けること。
なお、臨床試験では、eGFRが30mL/min/1.73m2未満の患者は除外されている。
なお、臨床試験では、eGFRが30mL/min/1.73m2未満の患者は除外されている。
9.3 肝機能障害患者
慎重な経過観察を行うこと。他の尿酸排泄促進薬では重篤な肝障害が認められている。
なお、臨床試験では、重篤な肝疾患を有する患者、AST又はALT100IU/L以上の患者は除外されている。[8.3参照]
なお、臨床試験では、重篤な肝疾患を有する患者、AST又はALT100IU/L以上の患者は除外されている。[8.3参照]
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。動物実験(ラット及びウサギ)で、臨床曝露量の約1053倍及び約174倍に相当する用量で骨格変異が認められた1)。
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。動物実験(ラット)で、本剤が乳汁中に移行することが報告されている2)。
10. 相互作用
10.2 併用注意
| ピラジナミド | 本剤の効果が減弱する可能性がある。 | ピラジナミドの代謝物がURAT1による尿酸再吸収を促進することが知られており、本剤の尿酸排泄促進に拮抗する可能性がある。 |
| サリチル酸製剤 アスピリン等 | 本剤の効果が減弱する可能性がある。 | サリチル酸製剤は尿酸の排泄を抑制することが知られており、本剤の尿酸排泄促進に拮抗する可能性がある。 |
11. 副作用
11.2 その他の副作用
| 5%以上 | 1〜5%未満 | 1%未満 | 頻度不明 | |
| 胃腸 | 軟便 | 下痢、悪心 | ||
| 肝及び胆道系 | γ-GTP増加 | ALT増加、AST増加 | ||
| 筋及び骨格系 | 痛風関節炎 | 関節炎、四肢不快感 | 関節痛 | |
| 腎及び泌尿器系 | 腎結石、腎石灰沈着症、尿中β2ミクログロブリン増加、血中クレアチニン増加、尿中アルブミン/クレアチニン比増加、尿中アルブミン陽性 | |||
| 皮膚 | 発疹、そう痒症 | |||
| その他 | 倦怠感 |
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
健康成人男性(36例)にドチヌラド0.5、1、2、5、10、20mgを絶食下で単回経口投与したときの血漿中未変化体濃度推移及び薬物動態学的パラメータを以下に示す。Cmax及びAUC0-infは用量依存的に増加し、線形性が認められた3)。
なお、本剤の承認された最大投与量は、ドチヌラドとして1回4mgを1日1回である。
なお、本剤の承認された最大投与量は、ドチヌラドとして1回4mgを1日1回である。
| 投与量 | Cmax(ng/mL) | Tmax(hr) | T1/2(hr) | AUC0-inf(ng・hr/mL) |
| 0.5mg(n=6) | 41.53±4.51 | 2.67±1.03 | 9.67±1.77 | 612.53±134.12 |
| 1mg(n=6) | 89.18±10.78 | 3.33±1.03 | 9.60±1.27 | 1276.01±189.17 |
| 2mg(n=5) | 175.22±33.01 | 3.10±1.24 | 9.53±1.11 | 2599.01±381.12 |
| 5mg(n=6) | 447.82±72.63 | 2.00±1.10 | 9.27±1.10 | 5525.68±419.02 |
| 10mg(n=6) | 858.18±136.26 | 3.25±1.17 | 9.87±0.83 | 12126.04±1204.32 |
| 20mg(n=6) | 1783.63±351.53 | 2.25±1.41 | 10.65±2.85 | 23397.97±7054.80 |
16.1.2 反復投与
健康成人男性(6例)にドチヌラド1回4mgを1日1回、摂食下で7日間反復経口投与したときの薬物動態学的パラメータを以下に示す。血漿中未変化体濃度は投与4日目には定常状態に達し、蓄積性は認められなかった4)。
| 投与日 | Cmax(ng/mL) | Tmax(hr) | T1/2(hr) | AUC0-24hr(ng・hr/mL) | 累積係数 |
| 1 | 366.50±81.19 | 3.33±0.52 | 11.14±1.56 | 4024.16±758.92 | − |
| 4 | 416.33±77.74 | 2.67±1.21 | 11.27±1.22 | 5052.31±1073.14 | − |
| 7 | 420.67±54.21 | 3.17±0.75 | 9.87±1.20 | 4871.26±890.21 | 0.97±0.07 |
16.2 吸収
16.2.1 食事の影響
健康成人男性(12例)にドチヌラド4mgを摂食下で単回経口投与したとき、絶食下投与と比較してCmaxは僅かに減少、Tmaxは延長したが、AUC0-tは食事の影響を受けなかった5)。
| 投与条件 | Cmax(ng/mL) | Tmax(hr) | T1/2(hr) | AUC0-t(ng・hr/mL) |
| 絶食下(n=12) | 296.48±37.26 | 2.58±0.87 | 9.35±0.89 | 3722.65±654.35 |
| 摂食下(n=11) | 261.59±52.19 | 3.91±1.51 | 9.05±1.09 | 3672.00±689.34 |
16.3 分布
16.3.1 分布容積
健康成人男性(6例)に14C-ドチヌラド1mgを絶食下で単回経口投与したときの分布容積は14.75Lであった6)。
16.3.2 蛋白結合率
ドチヌラドのヒト血漿蛋白結合率は99.2〜99.4%、ヒト血球移行は認められなかった7)(in vitro)。
16.4 代謝
ドチヌラドは主にUGT及びSULTによりグルクロン酸抱合体及び硫酸抱合体に代謝された。
健康成人男性(6例)に14C-ドチヌラド1mgを絶食下で単回経口投与したときの主な代謝物はグルクロン酸抱合体及び硫酸抱合体であった6)。
グルクロン酸抱合体生成にはUGT1A1、1A3、1A9及び2B7など、硫酸抱合体生成にはSULT1B1及び1A3など、それぞれ複数の分子種の関与が認められた8)(in vitro)。
ドチヌラドはCYP2C9に対し阻害作用を示した(Ki値10.4μmol/L)が、それ以外の分子種(CYP1A2、2A6、2B6、2C19、2D6、2E1及び3A4)はいずれも阻害しなかった(IC50>100μmol/L)9)。また、UGT1A1及び2B15に対し、阻害作用を示した(Ki値10.0及び16.6μmol/L)が、その他の分子種(UGT1A3、1A4、1A6、1A7、1A8、1A9、1A10、2B4、2B7、2B10及び2B17)はいずれも阻害しなかった(IC50>50μmol/L)10)(in vitro)。ドチヌラドはヒト肝細胞においてCYP2B6に対しmRNA発現誘導作用を示したが、CYP1A2及び3A4には誘導作用は示さなかった9)(in vitro)。これらの作用はいずれも臨床用量において相互作用が生じる可能性は低い。
16.5 排泄
健康成人男性(6例)に14C-ドチヌラド1mgを絶食下で単回経口投与したときの尿糞呼気中放射能排泄率は、投与後168時間までに尿中に投与量の86.38%、糞中に7.93%、投与後72時間までに呼気中に5.02%であった6)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害者
軽度及び中等度腎機能障害被験者(各6例)及び腎機能正常被験者(6例)にドチヌラド1mgを絶食下で単回経口投与したときの薬物動態パラメータは以下のとおりであった13)14)。
| 腎機能 | Cmax(ng/mL) | Tmax(hr) | T1/2(hr) | AUC0-inf(ng・hr/mL) |
| 正常(n=6) | 85.67±10.65 | 3.50±0.55 | 8.75±1.80 | 1157.32±269.46 |
| 軽度障害(n=6) | 88.73±22.74[1.01;0.79〜1.28] | 3.00±1.67 | 10.29±1.50 | 1366.57±427.94[1.15;0.84〜1.59] |
| 中等度障害(n=5) | 88.38±14.39[1.03;0.87〜1.21] | 2.60±0.55 | 10.95±2.17 | 1428.54±379.58[1.22;0.90〜1.66] |
16.6.2 肝機能障害者
軽度(6例)、中等度(9例)及び重度(3例)肝機能障害被験者及び肝機能正常被験者(6例)にドチヌラド4mgを絶食下で単回経口投与したときの薬物動態パラメータは以下のとおりであった15)16)。
| 肝機能 | Cmax(ng/mL) | Tmax(hr) | T1/2(hr) | AUC0-inf(ng・hr/mL) |
| 正常(n=6) | 339.15±28.57 | 2.67±1.03 | 10.80±0.55 | 4761.81±369.35 |
| 軽度障害(n=6) | 289.88±65.03[0.840;0.674〜1.047] | 2.17±1.17 | 10.50±2.42 | 4234.01±950.16[0.872;0.684〜1.112] |
| 中等度障害(n=9) | 280.34±87.91[0.798;0.653〜0.976] | 2.44±1.01 | 10.75±2.28 | 4327.09±1249.48[0.879;0.704〜1.098] |
| 重度障害(n=3) | 255.23±46.06[0.747;0.570〜0.979] | 1.33±0.58 | 9.82±2.47 | 3757.37±1343.74[0.758;0.563〜1.021] |
16.6.3 高齢者
非高齢者男性(20歳以上35歳以下の6例)及び高齢者男性(65歳以上の6例)、非高齢者女性(20歳以上35歳以下の6例)及び高齢者女性(65歳以上の6例)にドチヌラド1mgを絶食下で単回経口投与したときの薬物動態パラメータは以下のとおりであった17)18)。
| 投与群 | Cmax(ng/mL) | Tmax(hr) | T1/2(hr) | AUC0-inf(ng・hr/mL) | |
| 男性 | 高齢者(n=6) | 93.30±16.07[0.93;0.76〜1.15] | 2.00±0.63 | 9.28±1.05 | 1209.38±290.88[0.84;0.67〜1.06] |
| 非高齢者(n=6) | 100.92±21.20 | 2.17±0.75 | 10.31±1.27 | 1424.76±242.34 | |
| 女性 | 高齢者(n=6) | 112.07±12.66[0.98;0.80〜1.21] | 2.17±0.75 | 10.92±1.19 | 1797.95±357.84[0.98;0.80〜1.21] |
| 非高齢者(n=6) | 116.15±26.67 | 2.83±0.98 | 10.47±0.31 | 1832.67±345.74 | |
16.7 薬物相互作用
健康成人男性(12例)にドチヌラド4mgを摂食下で単回経口投与したとき、及びオキサプロジン600mgを1日1回、摂食下で5日間反復経口投与後、6日目にオキサプロジン600mg及びドチヌラド4mgを摂食下で経口併用投与したときのドチヌラドの薬物動態パラメータは以下のとおりであった19)20)。
| 投与群 | Cmax(ng/mL) | Tmax(hr) | T1/2(hr) | AUC0-inf(ng・hr/mL) |
| ドチヌラド単独(n=12) | 270.77±26.61 | 3.67±0.78 | 9.85±1.06 | 3845.95±578.70 |
| オキサプロジン併用(n=11) | 266.11±27.01[0.982;0.945〜1.021] | 3.64±0.81 | 11.89±1.33 | 4487.36±480.21[1.165;1.114〜1.219] |
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国内第III相試験(ベンズブロマロン対照)
痛風を含む高尿酸血症患者(尿酸産生過剰型を除く)201例を対象として、ベンズブロマロン対照二重盲検並行群間比較試験を行った。本剤を0.5mg/日から開始し、投与開始2週後に1mg/日、6週後に2mg/日へ段階的に増量し、以降、14週後まで用量を維持した。ベンズブロマロンは25mg/日から開始し、投与開始2週後に50mg/日へ増量し、14週後まで用量を維持した。
投与終了時における投与前値からの血清尿酸値低下率(主要評価項目)は下表のとおりであった。投与終了時の血清尿酸値低下率において、本剤2mg/日群のベンズブロマロン50mg/日群に対する非劣性が示された(非劣性マージンは−10%)。投与終了時の血清尿酸値6.0mg/dL以下の達成率(副次評価項目)は、本剤群が86.27%(88/102例)、ベンズブロマロン群が83.67%(82/98例)であった。
投与終了時における投与前値からの血清尿酸値低下率(主要評価項目)は下表のとおりであった。投与終了時の血清尿酸値低下率において、本剤2mg/日群のベンズブロマロン50mg/日群に対する非劣性が示された(非劣性マージンは−10%)。投与終了時の血清尿酸値6.0mg/dL以下の達成率(副次評価項目)は、本剤群が86.27%(88/102例)、ベンズブロマロン群が83.67%(82/98例)であった。
| 投与群 | 血清尿酸初期値(mg/dL) | 血清尿酸値低下率(%) | 低下率の群間差[95%信頼区間](%) |
| 本剤2mg/日(n=102) | 8.90±1.16 | 45.92±11.94 | 2.05[−1.27〜5.37] |
| ベンズブロマロン50mg/日(n=98) | 8.92±1.28 | 43.87±11.84 |
副作用の発現頻度は本剤群で14.7%(15/102例)、ベンズブロマロン群で15.2%(15/99例)であった。主な副作用は、本剤群では痛風関節炎7.8%(8/102例)、関節炎2.9%(3/102例)、ベンズブロマロン群では痛風関節炎5.1%(5/99例)、AST増加2.0%(2/99例)及びALT増加2.0%(2/99例)であった。各投与期間での痛風関節炎の発現率は下表のとおりであった21)22)。[5.、7.参照]
| 投与群 | 0〜2週以下 | 2週超6週以下 | 6週超14週以下 |
| 本剤2mg/日 | 1.0(1/102)(0.5mg/日) | 2.9(3/102)(1mg/日) | 4.0(4/100)(2mg/日) |
| ベンズブロマロン50mg/日 | 0.0(0/99)(25mg/日) | 2.0(2/99)(50mg/日) | 3.1(3/98)(50mg/日) |
17.1.2 国内第III相試験(フェブキソスタット対照)
痛風を含む高尿酸血症患者(尿酸産生過剰型を除く)201例を対象として、フェブキソスタット対照二重盲検並行群間比較試験を行った。本剤を0.5mg/日から開始し、投与開始2週後に1mg/日、6週後に2mg/日へ段階的に増量し、以降、14週後まで用量を維持した。フェブキソスタットは10mg/日から開始し、投与開始2週後に20mg/日、6週後に40mg/日へ増量し、14週後まで用量を維持した。投与終了時における投与前値からの血清尿酸値低下率(主要評価項目)は下表のとおりであった。投与終了時の血清尿酸値低下率において、本剤2mg/日群のフェブキソスタット40mg/日群に対する非劣性が示された(非劣性マージンは−10%)。投与終了時の血清尿酸値6.0mg/dL以下の達成率(副次評価項目)は、本剤群が84.8%(84/99例)、フェブキソスタット群が88.0%(88/100例)であった。
| 投与群 | 血清尿酸初期値(mg/dL) | 血清尿酸値低下率(%) | 低下率の群間差[95%信頼区間](%) |
| 本剤2mg/日(n=99) | 8.61±1.05 | 41.82±11.47 | −2.17[−5.26〜0.92] |
| フェブキソスタット40mg/日(n=100) | 8.67±1.06 | 44.00±10.63 |
副作用の発現頻度は本剤群で17.2%(17/99例)、フェブキソスタット群で19.8%(20/101例)であった。主な副作用は、フェブキソスタット群では痛風関節炎5.0%(5/101例)、尿中β2ミクログロブリン増加4.0%(4/101例)であった。なお、本剤群では、1%(1/99例)以下の副作用のみであった。各投与期間での痛風関節炎の発現率は下表のとおりであった23)24)。[5.、7.参照]
| 投与群 | 0〜2週以下 | 2週超6週以下 | 6週超14週以下 |
| 本剤2mg/日 | 0.0(0/99)(0.5mg/日) | 1.0(1/99)(1mg/日) | 1.0(1/97)(2mg/日) |
| フェブキソスタット40mg/日 | 0.0(0/101)(10mg/日) | 3.0(3/99)(20mg/日) | 2.1(2/96)(40mg/日) |
17.1.3 国内第III相長期投与試験
痛風を含む高尿酸血症患者(尿酸産生過剰型を除く)330例を対象として、長期投与試験を行った。本剤を0.5mg/日から開始し、投与開始2週後に1mg/日、6週後に2mg/日へ段階的に増量した。投与開始14週後に血清尿酸値が6.0mg/dLを超えていた場合は投与開始18週後から4mg/日へ増量し、以降34週後又は58週後まで用量を維持した。
投与前値からの血清尿酸値低下率は、34週間投与では2mg/日で46.73%、4mg/日で54.92%、58週間投与では2mg/日で47.17%、4mg/日で57.35%であった。血清尿酸値6.0mg/dL以下の達成率は、34週間投与では2mg/日で89.11%(229/257例)、4mg/日で97.50%(39/40例)、58週間投与では2mg/日で91.30%(84/92例)、4mg/日で100.00%(13/13例)であった。
副作用の発現頻度は21.8%(72/330例)であった。主な副作用は痛風関節炎12.7%(42/330例)、関節炎2.1%(7/330例)及び四肢不快感2.1%(7/330例)であった25)26)。[5.、7.参照]
投与前値からの血清尿酸値低下率は、34週間投与では2mg/日で46.73%、4mg/日で54.92%、58週間投与では2mg/日で47.17%、4mg/日で57.35%であった。血清尿酸値6.0mg/dL以下の達成率は、34週間投与では2mg/日で89.11%(229/257例)、4mg/日で97.50%(39/40例)、58週間投与では2mg/日で91.30%(84/92例)、4mg/日で100.00%(13/13例)であった。
副作用の発現頻度は21.8%(72/330例)であった。主な副作用は痛風関節炎12.7%(42/330例)、関節炎2.1%(7/330例)及び四肢不快感2.1%(7/330例)であった25)26)。[5.、7.参照]
18. 薬効薬理
18.1 作用機序
ドチヌラドは腎臓における尿酸の再吸収に関与するトランスポーターであるURAT1を選択的に阻害することにより、糸球体でろ過された尿酸の尿中排泄を促進し、血中尿酸値を低下させる。
18.2 URAT1阻害作用
18.3 血中尿酸値低下作用
19. 有効成分に関する理化学的知見
19.1. ドチヌラド
| 一般的名称 | ドチヌラド |
|---|---|
| 一般的名称(欧名) | Dotinurad |
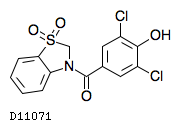
| 化学名 | (3,5-dichloro-4-hydroxyphenyl)(1,1-dioxo-1,2-dihydro-3H-1λ6-1,3-benzothiazol-3-yl)methanone |
| 分子式 | C14H9Cl2NO4S |
| 分子量 | 358.20 |
| 融点 | 約214℃ |
| 物理化学的性状 | 白色〜淡黄白色の結晶性の粉末である。本品はエタノール(99.5)又はメタノールに溶けにくく、水にほとんど溶けない。 |
| KEGG DRUG |
|
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
<ユリス錠0.5mg>
100錠[10錠(PTP)×10]、500錠[10錠(PTP)×50]
<ユリス錠1mg>
100錠[10錠(PTP)×10]、500錠[10錠(PTP)×50、バラ]
<ユリス錠2mg>
100錠[10錠(PTP)×10]
23. 主要文献
- 社内資料:生殖発生毒性試験(2020年1月23日承認,CTD 2.6.6.6)
- 社内資料:妊娠動物の組織分布(2020年1月23日承認,CTD 2.6.4.4)
- 社内資料:健康成人における単回投与試験(2020年1月23日承認,CTD 2.7.6.2)
- 社内資料:健康成人における反復投与試験(2020年1月23日承認,CTD 2.7.6.4)
- 社内資料:健康成人における食事の影響試験(2020年1月23日承認,CTD 2.7.6.1)
- 社内資料:マスバランス試験(2020年1月23日承認,CTD 2.7.6.5)
- 社内資料:血漿蛋白結合率及び血球移行性(2020年1月23日承認,CTD 2.7.2.2)
- 社内資料:UGT及びSULT分子種による代謝試験(2020年1月23日承認,CTD 2.7.2.2)
- 社内資料:CYP阻害及び誘導作用(2020年1月23日承認,CTD 2.7.2.2)
- 社内資料:UGT阻害作用(2020年1月23日承認,CTD 2.7.2.2)
- 社内資料:薬物トランスポーター阻害作用(2020年1月23日承認,CTD 2.7.2.2)
- Taniguchi T,et al., J Pharmacol Exp Ther., 371 (1), 162-170, (2019) »PubMed
- 社内資料:腎機能障害者における臨床薬理試験(2020年1月23日承認,CTD 2.7.6.6)
- Fukase H,et al., Clin Exp Nephrol., 24, S17-24, (2020)
- 社内資料:肝機能障害者における臨床薬理試験(2020年1月23日承認,CTD 2.7.6.7)
- Kumagai Y,et al., Clin Exp Nephrol., 24, S25-35, (2020)
- 社内資料:高齢者男女における臨床薬理試験(2020年1月23日承認,CTD 2.7.6.8)
- Nakatani H,et al., Clin Exp Nephrol., 24, S8-16, (2020)
- 社内資料:健康成人における薬物相互作用試験(2020年1月23日承認,CTD 2.7.6.9)
- Furihata K,et al., Clin Exp Nephrol., 24, S36-43, (2020)
- 社内資料:第III相ベンズブロマロン対照試験(2020年1月23日承認,CTD 2.7.6.15)
- Hosoya T,et al., Clin Exp Nephrol., 24, S62-70, (2020)
- 社内資料:第III相フェブキソスタット対照試験(2020年1月23日承認,CTD 2.7.6.16)
- Hosoya T,et al., Clin Exp Nephrol., 24, S71-79, (2020)
- 社内資料:第III相長期投与試験(2020年1月23日承認,CTD 2.7.6.17)
- Hosoya T,et al., Clin Exp Nephrol., 24, S80-91, (2020)
- 社内資料:ヒトURAT1発現細胞を用いた尿酸取り込み阻害試験(2020年1月23日承認,CTD 2.6.2.2)
- 社内資料:ヒトABCG2、OAT1及びOAT3発現細胞を用いた尿酸取り込み阻害試験(2020年1月23日承認,CTD 2.6.2.2)
- 社内資料:フサオマキザルを用いた血漿中尿酸値低下作用(2020年1月23日承認,CTD 2.6.2.2)
24. 文献請求先及び問い合わせ先
文献請求先
株式会社富士薬品
カスタマーサービスセンター
〒330-0854
埼玉県さいたま市大宮区桜木町1-12-5 沢田ビル6階
電話:048-644-3247 (受付時間 9:00〜17:30(土・日・祝日及び当社休日を除く))
FAX:048-644-2241
持田製薬株式会社
くすり相談窓口
〒160-8515
東京都新宿区四谷1丁目7番地
電話:03-5229-3906
0120-189-522
0120-189-522
FAX:03-5229-3955
製品情報問い合わせ先
株式会社富士薬品
カスタマーサービスセンター
〒330-0854
埼玉県さいたま市大宮区桜木町1-12-5 沢田ビル6階
電話:048-644-3247 (受付時間 9:00〜17:30(土・日・祝日及び当社休日を除く))
FAX:048-644-2241
持田製薬株式会社
くすり相談窓口
〒160-8515
東京都新宿区四谷1丁目7番地
電話:03-5229-3906
0120-189-522
0120-189-522
FAX:03-5229-3955
26. 製造販売業者等
26.1 製造販売元
株式会社富士薬品
埼玉県さいたま市大宮区桜木町4丁目383番地
26.2 販売
持田製薬株式会社
東京都新宿区四谷1丁目7番地