医薬品情報
| 総称名 | イブランス |
|---|---|
| 一般名 | パルボシクリブ |
| 欧文一般名 | Palbociclib |
| 製剤名 | パルボシクリブ錠 |
| 薬効分類名 | 抗悪性腫瘍剤(CDK4/6阻害剤) |
| 薬効分類番号 | 4291 |
| ATCコード | L01EF01 |
| KEGG DRUG |
D10372
パルボシクリブ
|
| KEGG DGROUP |
DG03138
CDK阻害薬
|
| JAPIC | 添付文書(PDF) |
添付文書情報2024年12月 改訂(第4版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| イブランス錠25mg | IBRANCE 25mg Tablets | ファイザー | 4291051F1022 | 5076.8円/錠 | 劇薬, 処方箋医薬品注) |
| イブランス錠125mg | IBRANCE 125mg Tablets | ファイザー | 4291051F2029 | 20538.9円/錠 | 劇薬, 処方箋医薬品注) |
1. 警告
1.1 本剤は、緊急時に十分対応できる医療施設において、がん化学療法に十分な知識・経験を持つ医師のもとで、本剤の投与が適切と判断される症例についてのみ投与すること。また、治療開始に先立ち、患者又はその家族に有効性及び危険性を十分説明し、同意を得てから投与すること。
2. 禁忌
次の患者には投与しないこと
2.1 本剤の成分に対し過敏症の既往歴のある患者
2.2 妊婦又は妊娠している可能性のある女性[9.5参照]
4. 効能または効果
ホルモン受容体陽性かつHER2陰性の手術不能又は再発乳癌
5. 効能または効果に関連する注意
本剤の術前・術後薬物療法としての有効性及び安全性は確立していない。
6. 用法及び用量
内分泌療法剤との併用において、通常、成人にはパルボシクリブとして1日1回125mgを3週間連続して経口投与し、その後1週間休薬する。これを1サイクルとして投与を繰り返す。なお、患者の状態により適宜減量する。
7. 用法及び用量に関連する注意
7.2 副作用があらわれた場合は、以下の基準を考慮して、休薬、減量又は投与を中止すること。なお、本剤は75mg/日未満に減量しないこと。
| 減量レベル | 投与量 |
| 通常投与量 | 125mg/日 |
| 一次減量 | 100mg/日 |
| 二次減量 | 75mg/日 |
| 副作用 | 処置 |
| Grade1又は2 | 同一投与量を継続する。 |
| Grade3 | 休薬し、1週間以内に血液検査(血球数算定)を行う。Grade2以下に回復後、同一投与量で投与を再開する。 Grade3の好中球減少の回復に日数を要する場合(1週間以上)や次サイクルでGrade3の好中球減少が再発する場合は、減量を考慮すること。 |
| Grade3 好中球減少に付随して38.5℃以上の発熱又は感染症がある場合 | Grade2以下に回復するまで休薬する。回復後、1レベル減量し投与を再開する。 |
| Grade4 | Grade2以下に回復するまで休薬する。回復後、1レベル減量し投与を再開する。 |
| 副作用 | 処置 |
| Grade1又は2 | 同一投与量を継続する。 |
| Grade3以上 治療しても症状が継続する場合 | Grade1以下又はGrade2で安全性に問題がない状態に回復するまで休薬する。 回復後、1レベル減量し投与を再開する。 |
8. 重要な基本的注意
8.1 骨髄抑制があらわれることがあるので、本剤の投与開始前及び投与中は定期的に血液検査を行い、患者の状態を十分に観察すること。[11.1.1参照]
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.3 肝機能障害患者
9.3.1 重度の肝機能障害のある患者
減量を考慮するとともに、患者の状態をより慎重に観察し、有害事象の発現に十分注意すること。本剤の血中濃度が上昇し、副作用が強くあらわれるおそれがある。[16.6.1参照]
9.4 生殖能を有する者
9.4.1 妊娠可能な女性に対しては、本剤の投与期間中及び治療終了から一定期間は適切な避妊を行うよう指導すること。[9.5参照]
9.5 妊婦
9.6 授乳婦
授乳しないことが望ましい。本剤のヒト乳汁中への移行については不明であるが、本剤はBCRPの基質であるため、乳汁移行の可能性がある。
9.7 小児等
小児等を対象とした臨床試験は実施していない。
9.8 高齢者
一般に生理機能が低下していることが多い。
10. 相互作用
相互作用序文
薬物代謝酵素用語
CYP3A
薬物代謝酵素用語
硫酸転移酵素(SULT)2A1
10.2 併用注意
| CYP3A阻害剤 コビシスタット、エルビテグラビル、インジナビル、イトラコナゾール、リトナビル、テラプレビル、ボリコナゾール、クラリスロマイシン、グレープフルーツジュース、ネルフィナビル、サキナビル等 [16.7.1参照] | 本剤の血中濃度が上昇し、副作用の発現頻度及び重症度が増加するおそれがあるので、CYP3A阻害作用のない薬剤への代替を考慮すること。 | これらの薬剤等がCYP3Aの代謝活性を阻害するため、本剤の血中濃度が上昇する可能性がある。 |
| 強いCYP3A誘導剤 フェニトイン、カルバマゼピン、リファンピシン、リファブチン、フェノバルビタール、セイヨウオトギリソウ含有食品等 [16.7.2参照] | 本剤の血中濃度が低下し、本剤の有効性が減弱するおそれがあるので、CYP3A誘導作用のない又は弱い薬剤への代替を考慮すること。 | これらの薬剤等がCYP3Aの代謝活性を誘導するため、本剤の血中濃度が低下する可能性がある。 |
| CYP3Aの基質となる薬剤 ミダゾラム、フェンタニル等 [16.7.3参照] | CYP3Aにより代謝される薬剤と併用する場合は、これらの薬剤の血中濃度が上昇する可能性がある。 | 本剤のCYP3A阻害作用により、これらの薬剤の代謝が阻害される。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 骨髄抑制
好中球減少(81.4%)、白血球減少(46.9%)、貧血(23.6%)、血小板減少(20.0%)、発熱性好中球減少症(1.4%)等があらわれることがある。[8.1参照]
11.1.2 間質性肺疾患(0.5%)
11.2 その他の副作用
| 20%以上 | 20%未満10%以上 | 10%未満 | 頻度不明 | |
| 皮膚 | 脱毛症 | 発疹 | 皮膚乾燥、手足症候群 | 多形紅斑 |
| 眼 | 流涙増加、霧視、眼乾燥 | |||
| 代謝 | 食欲減退 | |||
| 神経系 | 味覚異常 | |||
| 呼吸器 | 鼻出血 | |||
| 消化器 | 悪心、口内炎 | 下痢 | 嘔吐 | |
| 腎臓 | 腎機能障害(血中クレアチニン増加等) | |||
| その他 | 疲労 | 感染症(尿路感染、上気道感染、口腔ヘルペス、歯肉炎、上咽頭炎等) | 無力症、発熱、AST増加、ALT増加 |
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
15. その他の注意
15.2 非臨床試験に基づく情報
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
日本人健康成人14例にパルボシクリブ(カプセル剤)75、100、125及び150mg注1)を食後に単回投与したとき、投与後6時間でCmaxに達した。消失半減期は約23時間であり、Cmax及びAUCinfは用量に比例して増加した5)。
| 投与量(mg) | N | Cmax(ng/mL) | AUCinf(ng・h/mL) | Tmax(h) | t1/2(h) |
| 75 | 11 | 37.34(20) | 1071(24) | 6.00(6.00-8.02) | 23.4(16.1) |
| 100 | 11 | 51.79(18) | 1487(21) | 6.00(2.00-8.10) | 23.5(14.0) |
| 125 | 11 | 65.16(23) | 2021(20) | 8.00(4.02-12.0) | 23.3(13.2) |
| 150 | 11 | 86.64(26) | 2497(22) | 6.05(6.00-12.0) | 23.4(14.2) |
単回投与時の血漿中濃度推移(平均値+標準偏差)
16.1.2 反復投与
日本人進行乳癌患者6例にパルボシクリブ(カプセル剤)125mgを食後に反復投与したときのサイクル1第15日(定常状態時)の薬物動態パラメータを以下に示す。
| 投与量(mg) | N | Cmax(ng/mL) | AUC24h(ng・h/mL) | Tmax(h) | Ctrough(ng/mL) |
| 125 | 6 | 124.7(26) | 1979(16) | 4.90(2.00-8.20) | 59.75(38) |
反復投与時の血漿中濃度推移(平均値±標準偏差)
日本人進行固形癌患者6例にパルボシクリブ(カプセル剤)125mgを空腹時反復投与したときの累積係数は1.9であり、t1/2から予測される値と一致した6)。
16.2 吸収
16.2.1 バイオアベイラビリティ
健康成人14例にパルボシクリブ(カプセル剤)125mgを空腹時単回経口投与及びパルボシクリブ50mg注1)を単回静脈内投与注1)したときの絶対的バイオアベイラビリティは46%であった7)(外国人データ)。
16.2.2 食事の影響
健康成人44例に本剤125mgを単回経口投与したとき、AUClast及びCmaxは、空腹時と比較して高脂肪食注2)後にそれぞれ23%及び26%増加し、中程度の脂肪食注3)後に単回経口投与したときのAUClast及びCmaxは、それぞれ9%及び10%増加した8)(外国人データ)。
16.2.3 生物学的同等性
健康成人44例に本剤又はカプセル剤125mgを中程度の脂肪食注3)後に単回経口投与したとき、カプセル剤に対する本剤のAUClast及びCmaxの幾何平均値の比[90%信頼区間]は、それぞれ0.992[0.964,1.02]及び1.00[0.959,1.05]であり、いずれも生物学的同等性の判定基準範囲内(0.8〜1.25)であった9)(外国人データ)。
16.3 分布
健康成人14例にパルボシクリブ50mg注1)を空腹時単回静脈内投与注1)したとき、分布容積の平均値(変動係数)は1008L(29%)であった。癌患者にパルボシクリブ(カプセル剤)125mgを空腹時反復経口投与したときの定常状態時の見かけの分布容積(変動係数)は2583L(26%)であった。
In vitro試験より、パルボシクリブのヒト血漿蛋白結合率は約85%であり、蛋白結合率は500〜5000ng/mLの範囲では薬物濃度に依存しなかった10)(外国人データ)。
In vitro試験より、パルボシクリブのヒト血漿蛋白結合率は約85%であり、蛋白結合率は500〜5000ng/mLの範囲では薬物濃度に依存しなかった10)(外国人データ)。
16.4 代謝
16.5 排泄
健康成人6例に14Cで標識したパルボシクリブ125mgを空腹時単回経口投与したとき、投与後15日までに投与放射能の91.6%が回収され、投与放射能の74.1%が糞中に、17.5%が尿中に排泄された。未変化体の糞中及び尿中への排泄率は投与量のそれぞれ2.3%及び6.9%であり、パルボシクリブは主に代謝物として排泄された12)(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 肝機能障害患者
正常肝機能の被験者並びに軽度、中等度及び重度の肝機能障害を有する被験者28例に、パルボシクリブ(カプセル剤)75mg注1)を単回投与したときのパルボシクリブの非結合型濃度から求めたAUCinfは、正常肝機能の被験者と比較して、軽度の肝機能障害を有する被験者(Child-Pugh分類A)では17%減少し、中等度(Child-Pugh分類B)及び重度(Child-Pugh分類C)の肝機能障害を有する被験者ではそれぞれ34%及び77%増加した。また、パルボシクリブの非結合型濃度から求めたCmaxは、正常肝機能の被験者と比較して、軽度、中等度及び重度の肝機能障害を有する被験者では、それぞれ7%、38%及び72%増加した13)(外国人データ)。[9.3.1参照]
16.6.2 腎機能障害患者
正常腎機能の被験者並びに軽度、中等度及び重度の腎機能障害を有する被験者31例に、パルボシクリブ(カプセル剤)125mgを単回投与したときのパルボシクリブのAUCinfは、正常腎機能(クレアチニンクリアランス≧90mL/min)の被験者と比較して、軽度(60mL/min≦クレアチニンクリアランス<90mL/min)、中等度(30mL/min≦クレアチニンクリアランス<60mL/min)及び重度(クレアチニンクリアランス<30mL/min)の腎機能障害を有する被験者でそれぞれ、39%、42%及び31%増加した。パルボシクリブのCmaxは、正常腎機能の被験者と比較して、軽度、中等度及び重度の腎機能障害を有する被験者でそれぞれ17%、12%及び15%増加した14)(外国人データ)。なお、血液透析が必要な患者を対象とした試験は実施されていない。
16.7 薬物相互作用
16.7.1 イトラコナゾール
16.7.2 リファンピシン
16.7.3 ミダゾラム
16.7.4 その他
(1)健康成人14例にパルボシクリブ(カプセル剤)125mgを食後にモダフィニル(400mg反復投与)と併用投与したとき、単独投与時と比べ、パルボシクリブのAUCinf及びCmaxはそれぞれ32%及び11%減少した18)(外国人データ)。
(2)健康成人12例に本剤125mgを空腹時にラベプラゾール(40mg反復投与)と併用投与したとき、単独投与時と比べ、パルボシクリブのAUCinfは6%増加し、Cmaxは3%減少した19)(外国人データ)。
(3)健康成人25例にパルボシクリブ(カプセル剤)125mgを空腹時にタモキシフェン(20mg 1日1回反復投与)と併用投与したとき、単独投与時と比べ、パルボシクリブのAUCinf及びCmaxはそれぞれ13%及び20%増加した20)(外国人データ)。
(4)パルボシクリブはin vitro試験において、消化管のP-gp及びBCRP、並びにOCT1に対し阻害作用を示した21)。
注1)本剤の承認用法・用量は1日1回125mgを経口投与する。
注2)総カロリー約800〜1000kcalのうち脂質を約50%の割合で含む。
注3)総カロリー約500〜700kcalのうち脂質を約35%の割合で含む。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国際共同第III相試験
HR陽性かつHER2陰性であり、進行乳癌に対して内分泌療法歴のない手術不能又は再発閉経後乳癌患者666例(日本人46例を含む)を対象に、パルボシクリブ+レトロゾール併用投与とプラセボ+レトロゾール併用投与の有効性を検討することを目的とした、無作為化、二重盲検、並行群間、国際共同第III相試験を実施した。パルボシクリブは、開始用量としてカプセル剤125mgを1日1回3週間連続経口投与後1週間休薬し、レトロゾールは2.5mgを1日1回連続投与した。
主要評価項目である無増悪生存期間の中央値は、パルボシクリブ+レトロゾール群で24.8ヵ月、プラセボ+レトロゾール群で14.5ヵ月であり、ハザード比0.576(95%信頼区間:0.463,0.718;片側層別ログランク検定p<0.000001)でパルボシクリブ+レトロゾール群で統計学的に有意な無増悪生存期間の延長が認められた22)。
主要評価項目である無増悪生存期間の中央値は、パルボシクリブ+レトロゾール群で24.8ヵ月、プラセボ+レトロゾール群で14.5ヵ月であり、ハザード比0.576(95%信頼区間:0.463,0.718;片側層別ログランク検定p<0.000001)でパルボシクリブ+レトロゾール群で統計学的に有意な無増悪生存期間の延長が認められた22)。
治験責任医師判定に基づく無増悪生存期間のKaplan-Meier曲線(全解析対象集団)
パルボシクリブ(カプセル剤)が投与された444例(日本人32例を含む)中428例(96.4%)に副作用が認められた。主な副作用は、好中球減少症348例(78.4%)、白血球減少症171例(38.5%)、脱毛症140例(31.5%)、疲労134例(30.2%)、口内炎103例(23.2%)、悪心96例(21.6%)、関節痛87例(19.6%)、貧血85例(19.1%)、感染症85例(19.1%)、ほてり79例(17.8%)、下痢66例(14.9%)、血小板減少症65例(14.6%)、無力症55例(12.4%)、発疹48例(10.8%)等であった。[7.1参照]
17.1.2 国際共同第III相試験
HR陽性かつHER2陰性であり、内分泌療法に抵抗性の手術不能又は再発乳癌患者(閉経状態を問わない)521例(日本人35例を含む)を対象に、パルボシクリブ+フルベストラント併用投与とプラセボ+フルベストラント併用投与の有効性を検討することを目的とした、無作為化、二重盲検、並行群間、国際共同第III相試験を実施した。パルボシクリブは、開始用量としてカプセル剤125mgを1日1回3週間連続経口投与後1週間休薬し、フルベストラントは500mgを初回、2週後、4週後、その後4週ごとに投与した。閉経前・閉経周辺期患者にはゴセレリンを併用投与した。
中間解析時点(2014年12月5日カットオフ)において主要評価項目である無増悪生存期間の顕著な延長が認められ、事前に規定した中止基準を満たし、本試験は有効中止となった。無増悪生存期間の中央値は、パルボシクリブ+フルベストラント群で9.2ヵ月、プラセボ+フルベストラント群で3.8ヵ月であり、ハザード比0.422(95%信頼区間:0.318,0.560;片側層別ログランク検定p<0.000001)でパルボシクリブ+フルベストラント群で統計学的に有意な無増悪生存期間の延長が認められた23)。
中間解析時点(2014年12月5日カットオフ)において主要評価項目である無増悪生存期間の顕著な延長が認められ、事前に規定した中止基準を満たし、本試験は有効中止となった。無増悪生存期間の中央値は、パルボシクリブ+フルベストラント群で9.2ヵ月、プラセボ+フルベストラント群で3.8ヵ月であり、ハザード比0.422(95%信頼区間:0.318,0.560;片側層別ログランク検定p<0.000001)でパルボシクリブ+フルベストラント群で統計学的に有意な無増悪生存期間の延長が認められた23)。
治験責任医師判定に基づく無増悪生存期間のKaplan-Meier曲線(全解析対象集団)
パルボシクリブ(カプセル剤)が投与された345例(日本人27例を含む)中325例(94.2%)に副作用が認められた。主な副作用は、好中球減少症285例(82.6%)、白血球減少症198例(57.4%)、疲労114例(33.0%)、貧血96例(27.8%)、悪心87例(25.2%)、血小板減少症80例(23.2%)、口内炎70例(20.3%)、脱毛症57例(16.5%)、感染症50例(14.5%)、下痢45例(13.0%)、ほてり45例(13.0%)、発疹40例(11.6%)等であった(2016年2月26日カットオフ時点の集計)。[7.1参照]
17.1.3 国際共同第III相試験
HR陽性かつHER2陰性であり、進行乳癌に対して内分泌療法歴がない、又は進行乳癌に対する1レジメンの内分泌療法にて疾患進行が認められた手術不能又は再発乳癌患者(閉経状態を問わない)184例(日本人118例を含む)を対象に、パルボシクリブ+タモキシフェン併用投与とプラセボ+タモキシフェン併用投与の有効性を検討することを目的とした、無作為化、二重盲検、並行群間、国際共同第III相試験を日本を含むアジアで実施した。パルボシクリブは、開始用量としてカプセル剤125mgを1日1回3週間連続経口投与後1週間休薬し、タモキシフェンは20mgを1日1回連続投与した。閉経前・閉経周辺期患者にはゴセレリンを併用投与した。
主要評価項目である無増悪生存期間の中央値は、パルボシクリブ+タモキシフェン群で24.4ヵ月、プラセボ+タモキシフェン群で11.1ヵ月であり、ハザード比0.602(95%信頼区間:0.428,0.848;片側層別ログランク検定p=0.002)でパルボシクリブ+タモキシフェン群で統計学的に有意な無増悪生存期間の延長が認められた24)。
主要評価項目である無増悪生存期間の中央値は、パルボシクリブ+タモキシフェン群で24.4ヵ月、プラセボ+タモキシフェン群で11.1ヵ月であり、ハザード比0.602(95%信頼区間:0.428,0.848;片側層別ログランク検定p=0.002)でパルボシクリブ+タモキシフェン群で統計学的に有意な無増悪生存期間の延長が認められた24)。
治験責任医師判定に基づく無増悪生存期間のKaplan-Meier曲線(全解析対象集団)
パルボシクリブ(カプセル剤)が投与された91例(日本人69例を含む)中87例(95.6%)に副作用が認められた。主な副作用は、好中球減少症83例(91.2%)、白血球減少症44例(48.4%)、血小板減少症31例(34.1%)、口内炎30例(33.0%)、貧血27例(29.7%)、アスパラギン酸アミノトランスフェラーゼ増加15例(16.5%)、アラニンアミノトランスフェラーゼ増加14例(15.4%)、発疹14例(15.4%)等であった。[7.1参照]
18. 薬効薬理
18.1 作用機序
パルボシクリブはサイクリン依存性キナーゼ(CDK)4及び6に対して阻害活性を有する低分子化合物である。パルボシクリブは、CDK4/6とサイクリンDの複合体の活性を阻害し、網膜芽細胞腫(Rb)タンパクのリン酸化を阻害することにより、細胞周期の進行を停止し、腫瘍の増殖を抑制すると考えられている25)。
18.2 抗腫瘍効果
パルボシクリブは、ヒト乳癌由来T47D及びMCF7細胞株の増殖を抑制した。また、ヒト乳癌由来ZR-75-1細胞株を皮下移植した重症複合型免疫不全マウスにおいて、腫瘍の増殖を抑制した26)。
19. 有効成分に関する理化学的知見
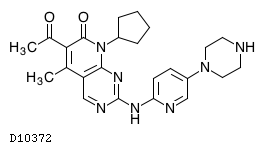
19.1. パルボシクリブ
| 一般的名称 | パルボシクリブ |
|---|---|
| 一般的名称(欧名) | Palbociclib |
| 化学名 | 6-Acetyl-8-cyclopentyl-5-methyl-2-{[5-(piperazin-1-yl)pyridin-2-yl]amino}pyrido[2,3-d]pyrimidin-7(8H)-one |
| 分子式 | C24H29N7O2 |
| 分子量 | 447.53 |
| 物理化学的性状 | パルボシクリブは黄色〜橙色の粉末である。N,N-ジメチルアセトアミドに溶けにくく、エタノール(99.5)及びメタノールに極めて溶けにくく、水にほとんど溶けない。 |
| 分配係数 | (log D):0.99(pH7.4、1-オクタノール/水) |
| KEGG DRUG |
|
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
<イブランス錠25mg>
30錠[10錠(PTP)×3]
<イブランス錠125mg>
7錠[7錠(PTP)×1]
23. 主要文献
- 社内資料:ウサギにおける生殖発生毒性試験(2017年9月27日承認、CTD2.6.6.6)
- 社内資料:ラット・イヌにおける反復投与毒性試験(2017年9月27日承認、CTD2.6.6.3及び2.6.6.9)
- 社内資料:遺伝毒性試験(2017年9月27日承認、CTD2.6.6.4)
- 社内資料:がん原性試験
- 社内資料:日本人健康成人における薬物動態(単回投与)(2017年9月27日承認、CTD2.7.2.2)
- 社内資料:日本人癌患者における薬物動態(反復投与)(2017年9月27日承認、CTD2.7.2.2)
- 社内資料:バイオアベイラビリティ(2017年9月27日承認、CTD2.7.1.2)
- 社内資料:食事の影響
- 社内資料:生物学的同等性
- 社内資料:分布(2017年9月27日承認、CTD2.7.2.3)
- 社内資料:代謝関連資料(2017年9月27日承認、CTD2.6.4.5)
- 社内資料:排泄経路(2017年9月27日承認、CTD2.7.2.2)
- 社内資料:肝機能障害を有する被験者における薬物動態(2017年9月27日承認、CTD2.7.2.2)
- 社内資料:腎機能障害を有する被験者における薬物動態(2017年9月27日承認、CTD2.7.2.2)
- 社内資料:イトラコナゾールとの薬物相互作用(2017年9月27日承認、CTD2.7.2.2)
- 社内資料:リファンピシンとの薬物相互作用(2017年9月27日承認、CTD2.7.2.2)
- 社内資料:ミダゾラムとの薬物相互作用(2017年9月27日承認、CTD2.7.2.2)
- 社内資料:モダフィニルとの薬物相互作用(2017年9月27日承認、CTD2.7.2.2)
- 社内資料:制酸薬の影響
- 社内資料:タモキシフェンとの薬物相互作用(2017年9月27日承認、CTD2.7.2.2)
- 社内資料:トランスポーター阻害能(2017年9月27日承認、CTD2.6.4.7)
- 社内資料:国際共同第III相試験(乳癌:PALOMA-2)(2017年9月27日承認、CTD2.7.6.20)
- 社内資料:国際共同第III相試験(乳癌:PALOMA-3)(2017年9月27日承認、CTD2.7.6.21)
- Kogawa T,et al., J Clin Oncol., 41 (no.17_suppl), LBA1068-LBA1068, (2023), (doi:10.1200/JCO.2023.41.17_suppl.LBA1068(乳癌:NCCH1607/PATHWAY))
- 社内資料:薬効薬理試験 作用機序(in vitro)(2017年9月27日承認、CTD2.6.2.2)
- 社内資料:薬効薬理試験 抗腫瘍効果(in vitro)(2017年9月27日承認、CTD2.6.2.2)
24. 文献請求先及び問い合わせ先
文献請求先
ファイザー株式会社
Pfizer Connect/メディカル・インフォメーション
〒151-8589
東京都渋谷区代々木3-22-7
電話:0120-664-467
製品情報問い合わせ先
ファイザー株式会社
Pfizer Connect/メディカル・インフォメーション
〒151-8589
東京都渋谷区代々木3-22-7
電話:0120-664-467
26. 製造販売業者等
26.1 製造販売元
ファイザー株式会社
東京都渋谷区代々木3-22-7