医薬品情報
| 総称名 | ベクルリー |
|---|---|
| 一般名 | レムデシビル |
| 欧文一般名 | Remdesivir |
| 製剤名 | レムデシビル・注射用凍結乾燥製剤 |
| 薬効分類名 | 抗ウイルス剤 |
| 薬効分類番号 | 6250 |
| ATCコード | J05AB16 |
| KEGG DRUG |
D11472
レムデシビル
|
| KEGG DGROUP |
DG03174
抗SARS-CoV-2薬
|
| JAPIC | 添付文書(PDF) |
添付文書情報2024年12月 改訂(第11版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ベクルリー点滴静注用100mg | VEKLURY for Intravenous Injection | ギリアド・サイエンシズ | 6250407D1020 | 46498円/瓶 | 注意−特例承認医薬品, 処方箋医薬品注) |
2. 禁忌
次の患者には投与しないこと
2.1 本剤の成分に対し過敏症の既往歴のある患者
4. 効能または効果
5. 効能または効果に関連する注意
6. 用法及び用量
通常、成人及び体重40kg以上の小児にはレムデシビルとして、投与初日に200mgを、投与2日目以降は100mgを1日1回点滴静注する。
通常、体重3.5kg以上40kg未満の小児にはレムデシビルとして、投与初日に5mg/kgを、投与2日目以降は2.5mg/kgを1日1回点滴静注する。
なお、総投与期間は10日までとする。
7. 用法及び用量に関連する注意
7.2 SARS-CoV-2による感染症の症状が発現してから速やかに投与を開始し、3日目まで投与する。ただし、SARS-CoV-2による肺炎を有する患者では、目安として、5日目まで投与し、症状の改善が認められない場合には10日目まで投与する。
8. 重要な基本的注意
8.1 肝機能障害があらわれることがあるので、投与前及び投与開始後は定期的に肝機能検査を行い、患者の状態を十分に観察すること。[11.1.1参照]
8.2 Infusion Reaction、アナフィラキシーを含む過敏症があらわれることがあるので、患者の状態を十分に観察するとともに、異常が認められた場合には直ちに投与を中止し、適切な処置を行うこと。また、これらの発現を回避できる可能性があるため、本剤の緩徐な投与を考慮すること。[7.1、11.1.2参照]
8.3 添加剤スルホブチルエーテルβ-シクロデキストリンナトリウムにより腎機能障害があらわれるおそれがあるので、投与前及び投与開始後は定期的に腎機能検査を行い、患者の状態を十分に観察すること。[9.2参照]
9. 特定の背景を有する患者に関する注意
9.2 腎機能障害患者
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。妊娠ラット及びウサギを用いた胚・胎児への影響に関する試験で、レムデシビル20mg/kgまでを静脈内投与した場合(主要血中代謝物(ヌクレオシド類似体)の全身曝露量(AUC)が国内承認用量投与時曝露量の4倍に相当)、胚・胎児発生に対する影響は認められなかった。雌ラットを用いた受胎能及び初期胚発生への影響に関する試験において、レムデシビル10mg/kgを静脈内投与した場合(主要血中代謝物(ヌクレオシド類似体)の全身曝露量(AUC)が国内承認用量投与時曝露量の1.3倍に相当)、黄体数・胚着床数・生存胚数の減少が認められている。
9.6 授乳婦
9.7 小児等
治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。28日齢未満の小児等を対象とした臨床試験結果は得られていない。[16.6.1参照]
9.8 高齢者
患者の状態を観察しながら慎重に投与すること。一般に生理機能が低下しており、既往歴や合併症を伴っていることが多くみられる。
10. 相互作用
相互作用序文
レムデシビルは有機アニオン輸送ポリペプチド(OATP)1B1の基質である。また、中間代謝物(GS-704277)はOATP1B1及びOATP1B3の基質である。[16.7.1参照]
薬物代謝酵素用語
OATP1B1
薬物代謝酵素用語
OATP1B3
10.2 併用注意
| ヒドロキシクロロキン硫酸塩 クロロキン(国内未承認) | レムデシビルの抗ウイルス活性が低下する可能性がある。 | レムデシビルの活性代謝物の生成及び抗ウイルス活性をクロロキンが阻害する可能性がある。 |
| シクロスポリン [16.7.2参照] | レムデシビル及び中間代謝物(GS-704277)の血漿中濃度が上昇するおそれがある。 | シクロスポリンの強力なOATP1B1/3阻害作用による。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 肝機能障害
ALT上昇に加えて、肝機能障害の徴候又は検査値異常(抱合型ビリルビン、ALP又はINRの異常)が認められた場合には、投与を中止すること。[8.1参照]
11.1.2 過敏症(Infusion Reaction、アナフィラキシーを含む)
低血圧、血圧上昇、頻脈、徐脈、低酸素症、発熱、呼吸困難、喘鳴、血管性浮腫、発疹、悪心、嘔吐、発汗、悪寒等があらわれることがある。[8.2参照]
11.2 その他の副作用
| 1%以上4%未満 | 0.1%以上1%未満 | 頻度不明 | |
| 血液およびリンパ系障害 | 貧血 | ||
| 心臓障害 | 徐脈 | ||
| 胃腸障害 | 悪心 | 嘔吐、便秘、下痢 | |
| 一般・全身障害および投与部位の状態 | 注入部位疼痛、疲労、発熱、悪寒 | ||
| 肝胆道系障害 | 高トランスアミナーゼ血症、高ビリルビン血症 | ||
| 臨床検査 | ALT増加、AST増加 | プロトロンビン時間延長、肝酵素上昇、肝機能検査値上昇、糸球体濾過率減少、血中クレアチニン増加、血中ビリルビン増加、トランスアミナーゼ上昇、ヘモグロビン減少 | |
| 代謝および栄養障害 | 高トリグリセリド血症 | ||
| 筋骨格系および結合組織障害 | 関節痛 | ||
| 神経系障害 | 頭痛、浮動性めまい | ||
| 精神障害 | 不眠症 | ||
| 皮膚および皮下組織障害 | 発疹、そう痒症、斑状皮疹 | ||
| 血管障害 | 静脈炎 |
14. 適用上の注意
14.1 薬剤調製時の注意
14.1.1 再溶解には、注射用水のみを用いること。
14.1.2 バイアルに19mLの注射用水を加え、直ちに30秒間撹拌し、2〜3分間静置した後、澄明な溶液であることを確認する(濃度5mg/mL)。内容物が溶解しきれない場合は、撹拌及び静置を繰り返す。
14.1.3 容器施栓系に欠陥・変色がなく、溶液中に微粒子がないことを目視で確認する。欠陥・変色や微粒子がみられた場合は使用しないこと。
14.1.4 成人及び体重40kg以上の小児については、初日の投与(レムデシビルとして200mg)の場合は、2バイアルを用い、各バイアルから20mLずつ(合計40mL)を、2日目以降(レムデシビルとして100mg)の投与の場合は、1バイアルから20mLをとり、生理食塩液に添加して全量を100mL又は250mLとする。体重3.5kg以上40kg未満の小児については、表1及び表2を参考に調製する。
14.1.5 静かに20回を目安に反転させて混和させるが、振とうは避けること。
14.1.6 注射用水で溶解してから、20〜25℃で24時間又は2〜8℃で48時間以内に使用すること15)。
| 体重(kg) | 初日の投与量(mg) | バイアル数 | 希釈後のバイアルから抜き取る量(mL) | 生理食塩液に添加後の全量(mL) |
| 3.5 | 17.5 | 1 | 3.5 | 25 |
| 4 | 20 | 1 | 4 | |
| 5 | 25 | 1 | 5 | |
| 7.5 | 37.5 | 1 | 7.5 | 50 |
| 10 | 50 | 1 | 10 | |
| 15 | 75 | 1 | 15 | 100 |
| 20 | 100 | 1 | 20 | |
| 25 | 125 | 2 | 25(20+5) | |
| 30 | 150 | 2 | 30(20+10) | |
| 35 | 175 | 2 | 35(20+15) | 250 |
| 体重(kg) | 体重40kg未満の小児における維持用量(mg) | バイアル数 | 希釈後のバイアルから抜き取る量(mL) | 生理食塩液に添加後の全量(mL) |
| 3.5 | 8.8 | 1 | 1.8 | 25 |
| 4 | 10 | 1 | 2 | |
| 5 | 12.5 | 1 | 2.5 | |
| 7.5 | 18.8 | 1 | 3.8 | 50 |
| 10 | 25 | 1 | 5 | |
| 15 | 37.5 | 1 | 7.5 | |
| 20 | 50 | 1 | 10 | |
| 25 | 62.5 | 1 | 12.5 | 100 |
| 30 | 75 | 1 | 15 | |
| 35 | 87.5 | 1 | 17.5 |
14.2 薬剤投与時の注意
14.2.1 他の薬剤と同時に投与しないこと。生理食塩液以外との適合性は不明である。
14.2.2 本剤は保存剤を含有しないため、調製後の未使用の希釈液及び使用後の残液は廃棄すること。
15. その他の注意
15.1 臨床使用に基づく情報
SARS-CoV-2による感染症患者を対象とした臨床試験(NIAID ACTT-1)では、プロトロンビン時間延長又は国際標準化比(INR)増加の発現割合はプラセボ群と比較して本剤投与群で高かった。なお、両投与群間で出血イベントの発現に差は認められなかった。
15.2 非臨床試験に基づく情報
アカゲザルを用いた7日間静脈内投与試験の20mg/kg/日群で腎毒性に伴う死亡、5mg/kg/日以上の群で血中尿素窒素・クレアチニンの増加等の腎機能障害、腎尿細管の組織傷害性、ラットを用いた14又は28日間静脈内投与試験において、臨床曝露量未満(10mg/kg/日以上)で血中腎機能マーカー異常・尿素窒素及びクレアチニンの増加、並びに尿中電解質・タンパク異常、腎尿細管の組織傷害性が認められた。なお、カニクイザルを用いた28日間静脈内投与試験で、最高用量10mg/kg/日群で腎毒性は認められていない。
16. 薬物動態
16.1 血中濃度
16.1.1 健康成人における薬物動態
外国人健康成人被験者に3mgから225mgの用量範囲でレムデシビルを2時間かけて単回静脈内投与したとき注)、レムデシビルは線形の薬物動態プロファイルを示した。
外国人健康被験者に、レムデシビルを投与初日は200mg、2〜5日目又は10日目に100mgを1日1回30分間かけて反復静脈内投与したときのレムデシビル、代謝物であるヌクレオシド類似体(GS-441524)及び中間代謝物(GS-704277)の薬物動態パラメータは以下のとおりであった。
外国人健康被験者に、レムデシビルを投与初日は200mg、2〜5日目又は10日目に100mgを1日1回30分間かけて反復静脈内投与したときのレムデシビル、代謝物であるヌクレオシド類似体(GS-441524)及び中間代謝物(GS-704277)の薬物動態パラメータは以下のとおりであった。
注)国内承認用法・用量は、投与初日に200mgを、投与2日目以降は100mgを1日1回点滴静注である。
| 用量(mg) | 例数 | 測定対象 | 測定日 | Cmax(ng/mL) | AUCa)(ng・h/mL) | t1/2b)(h) |
| 200 | 28 | レムデシビル | 1日目 | 4378(23.5) | 2863(18.6) | 0.90 |
| 100 | 26c) | 5日目及び10日目 | 2229(19.2) | 1585(16.6) | 0.96 | |
| 200 | 28 | ヌクレオシド類似体d) | 1日目 | 143(21.5) | 2191(19.1) | − |
| 100 | 26 | 5日目及び10日目 | 145(19.3) | 2229(18.4) | 27.4 | |
| 200 | 28 | 中間代謝物e) | 1日目 | 370(29.3) | 698(25.9) | 1.27 |
| 100 | 26 | 5日目及び10日目 | 246(33.9) | 462(31.4) | 1.23 |
16.1.2 患者における薬物動態
健康成人被験者並びに成人及び小児のSARS-CoV-2による感染症患者を対象とした試験の併合データを用いて構築した母集団薬物動態モデルにより推定した、成人患者(147例)での本剤の静脈内反復投与後のレムデシビル及びその循環血中代謝物[ヌクレオシド類似体(GS-441524)及び中間代謝物(GS-704277)]の薬物動態パラメータは以下のとおりであった(外国人のデータ)。
| レムデシビル | ヌクレオシド類似体b) | 中間代謝物c) | |
| Cmax(ng/mL) | 2700(2440-2990) | 143(135-152) | 198(180-218) |
| AUCtau(h*ng/mL) | 1710(1480-1980) | 2410(2250-2580) | 392(348-442) |
16.3 分布
In vitro試験において、レムデシビルのヒト血漿蛋白に対する結合率は88〜93%であった。ヌクレオシド類似体(GS-441524)のヒト血漿蛋白に対する結合率は低かった(2%)。
外国人健康成人に14C標識したレムデシビル150mgを単回静脈内投与したとき注)、総放射能の血液/血漿比は投与開始15分後で約0.68であり、時間の経過とともに上昇し、投与5時間後では1.0であった。レムデシビル及び代謝物は、血漿又は血液中の細胞成分に対して異なる分布を示す。
16.4 代謝
レムデシビルは主にカルボキシルエステラーゼ1(CES1)により加水分解され、一部カテプシンA(CatA)やCYP3Aにより代謝される。加水分解により生成された中間代謝物(GS-704277)は主にヒスチジントライアドヌクレオチド結合タンパク質1(HINT1)により代謝される。中間代謝物はホスホルアミダートの分解とそれに続くリン酸化により活性型三リン酸(GS-443902)となる。一方、脱リン酸化により、効率的に再リン酸化されないヌクレオシド代謝物(GS-441524)が生成される。
16.5 排泄
外国人健康成人被験者に14C標識レムデシビル150mgを単回静脈内投与したとき注)、投与量の平均総回収率は92%を超え、尿中及び糞中排泄率はそれぞれ約74%及び約18%であった。尿中に回収された大部分は、代謝物であるヌクレオシド類似体(GS-441524、49%)であり、10%がレムデシビルであった。
注)国内承認用法・用量は、投与初日に200mgを、投与2日目以降は100mgを1日1回点滴静注である。
16.6 特定の背景を有する患者
16.6.1 小児患者
健康成人被験者並びに成人及び小児のSARS-CoV-2による感染症患者を対象とした試験の併合データを用いて構築した母集団薬物動態モデルにより推定した、GS-US-540-5823試験における体重3.0kg以上の28日齢以上18歳未満の小児患者(50例)での本剤の静脈内反復投与後のレムデシビル及び代謝物(ヌクレオシド類似体[GS-441524]及び中間代謝物[GS-704277])の薬物動態パラメータは以下のとおりであった(外国人のデータ)。[9.7参照]
| コホート1: 12歳以上18歳未満かつ体重40kg以上(12例) | コホート2: 28日齢以上18歳未満かつ体重20kg以上40kg未満(12例) | コホート3: 28日齢以上18歳未満かつ体重12kg以上20kg未満(11例) | コホート4: 28日齢以上18歳未満かつ体重3kg以上12kg未満(10例) | コホート8: 12歳未満かつ体重40kg以上(5例) | |
| レムデシビル | |||||
| Cmax(ng/mL) | 3910(3140-4870) | 5680(4660-6930) | 5530(4240-7210) | 4900(3790-6340) | 3920(2270-6790) |
| AUCtau(h*ng/mL) | 2470(1940-3150) | 3500(2570-4780) | 3910(2140-7160) | 2930(1900-4520) | 2280(1200-4300) |
| ヌクレオシド類似体b) | |||||
| Cmax(ng/mL) | 197(123-316) | 181(132-248) | 158(116-215) | 202(171-238) | 162(57.4-458) |
| AUCtau(h*ng/mL) | 3460(2010-5960) | 2870(2020-4080) | 2400(1740-3320) | 2770(2230-3450) | 2640(772-9030) |
| 中間代謝物c) | |||||
| Cmax(ng/mL) | 307(212-443) | 423(309-578) | 444(336-585) | 390(305-500) | 278(145-532) |
| AUCtau(h*ng/mL) | 815(474-1400) | 754(547-1040) | 734(513-1050) | 691(494-966) | 537(203-1420) |
注)国内では、体重3.5kg以上の小児に対する用法・用量が承認されている。
16.6.2 腎機能障害
腎機能正常被験者に対する腎機能障害被験者(軽度、中等度、重度の腎機能障害及び末期腎不全)における単回投与時のレムデシビル及び代謝物(ヌクレオシド類似体[GS-441524]及び中間代謝物[GS-704277])の薬物動態パラメータ比は以下のとおりであった(外国人のデータ)。
| eGFRb)区分(mL/min/1.73m2) | ||||||
| 60以上90未満 (10例) | 30以上60未満 (10例) | 15以上30未満 (10例) | 15未満 | |||
| 血液透析前 (6例) | 血液透析後 (6例) | 血液透析なし (3例) | ||||
| レムデシビル | ||||||
| Cmax | 0.96(0.71-1.31) | 1.20(1.01-1.42) | 0.97(0.83-1.13) | 0.89(0.67-1.18) | 1.13(0.79-1.60) | 0.94(0.65-1.35) |
| AUCinf | 1.00(0.75-1.32) | 1.22(0.98-1.52) | 0.94(0.83-1.07) | 0.80c)(0.59-1.08) | 1.08c)(0.72-1.63) | 0.89(0.55-1.43) |
| ヌクレオシド類似体d) | ||||||
| Cmax | 1.07(0.90-1.26) | 1.44(1.12-1.85) | 1.68(1.28-2.20) | 2.26(1.72-2.99) | 3.07(2.21-4.26) | 3.00(2.63-3.42) |
| AUCinf | 1.19(0.97-1.47) | 2.02(1.57-2.62) | 3.26(2.39-4.46) | 4.97c)(3.65-6.77) | 6.22c)(4.44-8.71) | 7.87(6.49-9.53) |
| 中間代謝物e) | ||||||
| Cmax | 2.25(1.20-4.20) | 1.83(1.34-2.49) | 1.27(0.96-1.68) | 1.43(1.00-2.05) | 1.23(0.84-1.80) | 1.76(1.18-2.61) |
| AUCinf | 1.39(1.13-1.71) | 2.01(1.48-2.73) | 1.78(1.27-2.49) | 2.18c)(1.61-2.95) | 2.06c)(1.42-2.97) | 2.81(1.79-4.43) |
16.6.3 肝機能障害
肝機能正常被験者に対する中等度又は重度(Child-Pugh分類B又はC)肝機能障害被験者における単回投与時のレムデシビル及び代謝物(ヌクレオシド類似体[GS-441524]及び中間代謝物[GS-704277])の薬物動態パラメータ比は以下のとおりであった(外国人のデータ)22)。
| 中等度肝機能障害 (10例) | 重度肝機能障害 (6例) | |
| レムデシビル | ||
| Cmax | 1.10(0.75-1.60) | 1.03(0.70-1.51) |
| AUCinf | 1.21(0.87-1.67) | 1.56(1.20-2.03) |
| ヌクレオシド類似体b) | ||
| Cmax | 1.09(0.86-1.38) | 1.48(1.17-1.86) |
| AUCinf | 0.90(0.69-1.17) | 1.31(0.93-1.84) |
| 中間代謝物c) | ||
| Cmax | 1.29(0.54-3.08) | 1.07(0.70-1.63) |
| AUCinf | 1.38(0.92-2.07) | 2.41(1.70-3.42) |
16.6.4 妊婦
妊娠していない女性(22例)に対する妊婦(21例)でのレムデシビル及び代謝物(ヌクレオシド類似体[GS-441524]及び中間代謝物[GS-704277])の薬物動態パラメータ比は以下のとおりであった(外国人のデータ)19)。
| レムデシビル | ヌクレオシド類似体b) | 中間代謝物c) | |
| Cmax | 1.1(0.7-1.7) | 0.9(0.8-1.1) | 1.0(0.9-1.2) |
| AUCtau | 1.0(0.7-1.4) | 0.9(0.7-1.1) | 1.0(0.9-1.2) |
16.7 薬物相互作用
16.7.1 In vitro試験成績
レムデシビルはOATP1B1及びP-gpの基質である。また、CYP3A、UGT1A1、OATP1B1、OATP1B3及びMATE1に対し阻害作用を示す。中間代謝物(GS-704277)はOATP1B1及びOATP1B3の基質である。[10.参照]
16.7.2 臨床における薬物相互作用試験
薬物相互作用試験の結果を以下に示す(外国人のデータ)。[10.2参照]
| 併用薬 | 併用薬の投与量 | レムデシビルの投与量 | 例数 | レムデシビル及び代謝物の薬物動態パラメータ比(90%信頼区間) | ||
| Cmax | AUCinf | |||||
| シクロスポリン14) | 400mg 単回 | 100mg 単回 | 9 | レムデシビル | 1.49(1.38-1.60) | 1.89(1.77-2.02) |
| ヌクレオシド類似体a) | 1.17(1.12-1.22) | 1.03(0.99-1.08) | ||||
| 中間代謝物b) | 2.51(2.26-2.78) | 2.97(2.75-3.20) | ||||
| カルバマゼピン14) | 300mg 1日2回 | 100mg 単回 | 8 | レムデシビル | 0.87(0.78-0.97) | 0.92(0.83-1.02) |
| ヌクレオシド類似体a) | 0.97(0.88-1.07) | 0.83(0.78-0.89) | ||||
| 中間代謝物b) | 0.96(0.84-1.10) | 0.98(0.92-1.05) | ||||
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国際共同第III相試験
(1)NIAID ACTT-1試験(NCT04280705)
18歳以上のSARS-CoV-2による感染症患者(1,062例、うち15例は国内試験実施施設において登録された)を対象としたプラセボ対照無作為化二重盲検並行群間比較試験において、投与初日に本剤200mgを、2〜10日目に本剤100mgを1日1回、又はプラセボを静脈内投与した3)。なお、退院した場合は治験薬投与を中止することとされた。治験薬投与に加えて各国のSARS-CoV-2による感染症治療に関するガイドライン等に従った標準療法の実施が可能とされた。主要評価項目は、無作為化後28日目までにおける回復(8点順序尺度注1)のスコア1〜3に該当)までの時間であった。その結果、回復までの時間(中央値)について、本剤投与群で10日、プラセボ群で15日であり、本剤群とプラセボ群との対比較において統計学的に有意な差が認められた(ハザード比:1.29、95%信頼区間:1.12〜1.49、p<0.001、層別ログランク検定)。
図1 無作為化から回復までの時間のイベント発現割合
なお、本試験の主な選択・除外基準は下表のとおりであった。
| 選択基準 | 1.SARS-CoV-2による感染症が示唆される症状で入院中 2.以下のいずれかに該当しており、PCR等によりSARS-CoV-2感染が確認されている ・無作為化前72時間未満に採取された検体においてPCR陽性 ・無作為化前72時間以前に採取された検体においてPCR陽性で、追加の検体採取が困難であることが記録されている、かつSARS-CoV-2による感染によると思われる症状が進行している 3.少なくとも以下のいずれか1つが認められる患者 ・肺炎画像所見(胸部X線、CTスキャン等) ・SpO2が94%(室内気)以下 ・酸素吸入を要する ・人工呼吸器管理 |
| 除外基準 | 1.AST又はALTが基準範囲上限の5倍超 2.推定糸球体ろ過量(eGFR)が30mL/min未満(血液透析又は血液ろ過を受けている患者を含む) 3.妊婦又は授乳婦 4.72時間以内に退院又は転院予定 |
副作用注2)が認められた被験者の割合は、本剤投与群で8%(41/532例)であり、主な副作用はプロトロンビン時間延長2%(9/532例)であった。
注1)8点順序尺度[スコア1:退院かつ活動に制限なし、スコア2:退院かつ活動が制限及び/又は在宅酸素吸入が必要、スコア3:入院しており酸素吸入を要しない−治療の継続が不要、スコア4:入院、酸素吸入を要しない−治療の継続が必要(COVID-19関連又はそれ以外)、スコア5:入院かつ、酸素吸入を要する、スコア6:入院かつ非侵襲的人工呼吸器又は高流量酸素による管理、スコア7:入院かつECMO又は侵襲的人工呼吸器による管理、スコア8:死亡]
注2)本試験では、Grade3以上の有害事象が収集され、治験薬との因果関係が評価された。加えて、過敏症反応についてはGrade2の治験薬との因果関係が否定できない事象も収集された。
(2)GS-US-540-5773試験(NCT04292899)
12歳以上18歳未満かつ体重40kg以上、及び18歳以上の重症のSARS-CoV-2による感染症患者(397例、なお、日本人被験者は組み入れられなかった)を対象とした無作為化非盲検並行群間比較パートにおいて、5日間投与群では、投与初日に本剤200mgを、2〜5日目に100mgを1日1回静脈内投与、10日間投与群では、投与初日に本剤200mgを、2〜10日目に100mgを1日1回静脈内投与した4)5)。なお、退院した場合は治験薬投与を中止することとされた。いずれの投与群も標準療法の併用を受けた。主要評価項目は、無作為化後13日目に7点順序尺度注3)で評価した臨床状態とされた。臨床状態の改善について、5日間投与群に対する10日間投与群の比例オッズ比は0.75[95%信頼区間0.51,1.12]であった。
| スコア | 5日間投与群(200例) | 10日間投与群(197例) |
| 1 | 16(8.0) | 21(10.7) |
| 2 | 17(8.5) | 33(16.8) |
| 3 | 8(4.0) | 10(5.1) |
| 4 | 19(9.5) | 15(7.6) |
| 5 | 12(6.0) | 12(6.1) |
| 6 | 8(4.0) | 3(1.5) |
| 7 | 120(60.0) | 103(52.3) |
| 比例オッズ比 [95%信頼区間]a) | 0.75 [0.51,1.12] | |
なお、本試験の主な選択・除外基準は下表のとおりであった。
| 選択基準 | 1.無作為化前4日以内に実施したPCR検査においてSARS-CoV-2感染が確認されている 2.入院中 3.スクリーニング時に、SpO2が94%以下(室内気)又は酸素吸入を要する 4.画像上、肺浸潤影が認められる |
| 除外基準 | 1.多臓器不全 2.人工呼吸器(V-V ECMOを含む)を5日間以上使用、又はV-A ECMOを使用(使用期間を問わない) 3.ALT又はASTが基準範囲上限の5倍超 4.クレアチニン・クリアランスが50mL/min未満(18歳以上の場合はCockcroft-Gault式、18歳未満の場合はSchwartz式を用いて算出) 5.妊娠検査陽性 6.授乳中 |
副作用が認められた被験者の割合は、5日間投与群及び10日間投与群でそれぞれ17%(33/200例)及び20%(40/197例)であった。主な副作用は、ALT増加(5日間投与群で2%(4/200例)、10日間投与群で7%(14/197例))、AST増加(5日間投与群で3%(5/200例)、10日間投与群で6%(11/197例))及び悪心(5日間投与群で5%(9/200例)、10日間投与群で3%(5/197例))であった。
(3)GS-US-540-5774試験(NCT04292730)
12歳以上18歳未満かつ体重40kg以上、及び18歳以上の中等症のSARS-CoV-2による感染症患者(584例、なお、日本人被験者は組み入れられなかった)を対象とした無作為化非盲検並行群間比較パートにおいて、5日間投与群では、投与初日に本剤200mgを、2〜5日目に100mgを1日1回静脈内投与、10日間投与群では、投与初日に本剤200mgを、2〜10日目に100mgを1日1回静脈内投与し、標準療法群と比較した6)7)。なお、退院した場合は治験薬投与を中止することとされた。いずれの本剤投与群も標準療法の併用を受けた。主要評価項目は、無作為化後10日目に7点順序尺度注3)で評価した臨床状態とされた。臨床状態の改善について、比例オッズモデルに基づく標準療法群に対する各本剤投与群の比例オッズ比[95%信頼区間]は、5日間投与群で1.65[1.09,2.48,p=0.017]、10日間投与群では1.31[0.88,1.95,p=0.18]であった。
| スコア | 5日間投与群(191例) | 10日間投与群(193例) | SOC群(200例) |
| 1 | 0 | 2(1.0) | 4(2.0) |
| 2 | 0 | 1(0.5) | 4(2.0) |
| 3 | 5(2.6) | 0 | 7(3.5) |
| 4 | 7(3.7) | 12(6.2) | 11(5.5) |
| 5 | 38(19.9) | 44(22.8) | 46(23.0) |
| 6 | 7(3.7) | 9(4.7) | 8(4.0) |
| 7 | 134(70.2) | 125(64.8) | 120(60.0) |
| SOC群に対する比例オッズ比 [95%信頼区間]a) | 1.65 [1.092,2.483] | 1.31 [0.880,1.952] | − |
| p値b) | 0.0174 | 0.1826 |
なお、本試験の主な選択・除外基準は下表のとおりであった。
| 選択基準 | 1.無作為化前4日以内に実施したPCR検査においてSARS-CoV-2感染が確認されている 2.入院中であり、COVID-19に対する治療を要する 3.スクリーニング時に、SpO2が94%超(室内気) 4.画像上、肺浸潤影が認められる |
| 除外基準 | 1.スクリーニング時に人工呼吸器の使用を要する 2.ALT又はASTが基準範囲上限の5倍超 3.クレアチニン・クリアランスが50mL/min未満(18歳以上の場合はCockcroft-Gault式、18歳未満の場合はSchwartz式を用いて算出) 4.妊娠検査陽性 5.授乳中 |
副作用が認められた被験者の割合は、5日間投与群及び10日間投与群でそれぞれ19%(36/191例)及び13%(25/193例)であった。主な副作用は、悪心(5日間投与群で7%(13/191例)、10日間投与群で4%(7/193例))であった。
17.1.2 海外第III相試験
GS-US-540-9012試験(NCT04501952)
12歳以上18歳未満かつ体重40kg以上又は18歳以上の重症化リスク因子を一つ以上有するSARS-CoV-2による感染症患者(562例)を対象としたプラセボ対照無作為化二重盲検並行群間比較試験において、投与初日に本剤200mgを、2及び3日目に本剤100mgを1日1回、又はプラセボを静脈内投与した13)。治験薬投与に加えて各国のSARS-CoV-2による感染症治療に関するガイドライン等に従った標準療法の実施が可能とされた。主要評価項目は、無作為化後28日目までのSARS-CoV-2による感染症に伴う入院又は死因を問わない死亡(イベント)が認められた被験者の割合とされた。その結果、イベントの発現割合は、本剤投与群で0.7%(2/279例)、プラセボ群で5.3%(15/283例)であり、本剤投与群における無作為化後28日目までのSARS-CoV-2による感染症に伴う入院又は死因を問わない死亡が認められた被験者の割合は、プラセボ群と比較して87%低下した(ハザード比:0.134、95%信頼区間:0.031〜0.586、p=0.0076)。いずれの投与群でも無作為化後28日目までに死亡は認められなかった。
| 本剤投与群(279例) | プラセボ群(283例) | ||
| イベント発現 | 2(0.7) | 15(5.3) | |
| 入院 | 2(0.7) | 15(5.3) | |
| 死因を問わない死亡 | 0 | 0 | |
| プラセボ群に対するイベント発現低下率 | 87% | ||
| ハザード比a),b)[95%信頼区間]b) | 0.134[0.031,0.586] | ||
| p値b) | 0.0076 | ||
なお、本試験の主な選択・除外基準は下表のとおりであった。
| 選択基準 | 1.18歳以上又は12歳以上18歳未満かつ体重40kg以上で、次のSARS-CoV-2による感染症の重症化リスク因子を少なくとも一つ有する ・慢性肺疾患(慢性閉塞性肺疾患、中等症から重症の喘息、嚢胞性線維症、肺線維症) ・高血圧(全身性又は肺性) ・心血管系又は脳血管系疾患(冠動脈疾患、先天性心疾患、心不全、心筋症、脳卒中歴、心房細動、高脂血症) ・糖尿病(1型、2型、妊娠中) ・肥満(BMI30kg/m2以上) ・免疫不全状態(臓器移植、血液移植、又は骨髄移植の既往歴あり)、免疫不全(HIVによりCD4細胞数が低値又はHIVに対して未治療)、コルチコステロイドの長期使用又は他の免疫抑制薬の使用 ・軽度又は中等度の慢性腎臓病 ・慢性肝疾患 ・進行がん ・鎌状赤血球症 又は、60歳以上で重症化リスク因子の有無を問わない 2.スクリーニング前4日以内に実施した分子診断(核酸検査[例:PCR]又は抗原検査)においてSARS-CoV-2感染が確認されている 3.ランダム化前7日以内にCOVID-19による症状(発熱、咳、疲労、息切れ、咽喉痛、頭痛、筋肉痛・関節痛など)が少なくとも一つ確認されている 4.酸素吸入を要しない 5.入院(24時間以上の急性期治療)を要しない |
| 除外基準 | 1.COVID-19による入院(24時間以上の急性期治療)歴 2.SARS-CoV-2に直接作用する抗ウイルス活性を有する又はその可能性がある他の薬剤を投与、若しくはSARS-CoV-2(又はCOVID-19)ワクチンを接種 3.スクリーニング時又はスクリーニング前90日以内のALT又はASTが基準範囲上限の5倍超 4.18歳以上の場合は、スクリーニング時又はスクリーニング前90日以内のクレアチニン・クリアランスが30mL/min未満(Cockcroft-Gault式を用いて算出)、18歳未満の場合は、スクリーニング時又はスクリーニング前90日以内に推定糸球体ろ過量(eGFR)が30mL/min/1.73m2未満(Schwartz式を用いて算出) 5.授乳中 |
副作用が認められた被験者の割合は、本剤投与群で12.2%(34/279例)であり、主な副作用は悪心6.5%(18/279例)及び悪寒2.2%(6/279例)であった。
17.1.3 海外第II/III相試験
GS-US-540-5823試験(NCT04431453)
28日齢以上18歳未満のSARS-CoV-2による感染症患者(53例)を対象とした単群非盲検試験(12歳以上かつ体重40kg以上[12例]、12歳未満かつ体重40kg以上[5例]、28日齢以上かつ体重20kg以上40kg未満[12例]、28日齢以上かつ体重12kg以上20kg未満[12例]、28日齢以上かつ体重3kg以上12kg未満[12例])において、体重40kg以上の被験者では投与初日に本剤200mgを、以降最長10日目まで本剤100mgを1日1回静脈内投与し、体重3kg以上40kg未満の被験者では投与初日に本剤5mg/kgを、以降最長10日目まで2.5mg/kgを1日1回静脈内投与した18)。
本剤を最長10日間静脈内投与した結果、10日目におけるベースラインからの臨床状態の変化の中央値(第1四分位[Q1]、第3四分位[Q3])は、7段階の順序尺度注3)で2.0(1.0,4.0)ポイントの改善であった。62%の患者が10日目までに回復注4)し、回復までの期間の中央値(Q1、Q3)は7(5、16)日であった。また、60%の患者が10日目までに退院した。3名の被験者が本試験期間中に死亡した。
なお、本試験の主な選択・除外基準は下表のとおりであった。
本剤を最長10日間静脈内投与した結果、10日目におけるベースラインからの臨床状態の変化の中央値(第1四分位[Q1]、第3四分位[Q3])は、7段階の順序尺度注3)で2.0(1.0,4.0)ポイントの改善であった。62%の患者が10日目までに回復注4)し、回復までの期間の中央値(Q1、Q3)は7(5、16)日であった。また、60%の患者が10日目までに退院した。3名の被験者が本試験期間中に死亡した。
なお、本試験の主な選択・除外基準は下表のとおりであった。
| 選択基準 | 1.18歳未満で以下の体重基準のいずれかを満たす ・12歳以上18歳未満かつスクリーニング時の体重40kg以上 ・28日齢以上18歳未満かつスクリーニング時の体重3kg以上40kg未満 ・12歳未満かつスクリーニング時の体重40kg以上 2.PCR検査においてSARS-CoV-2感染が確認されている 3.入院中であり、COVID-19に対する治療を要する |
| 除外基準 | 1.治験薬投与前24時間以内にSARS-CoV-2に直接作用する抗ウイルス活性を有する又はその可能性がある他の薬剤を投与 2.ALT又はASTが基準範囲上限の5倍超 3.1歳以上の場合は、eGFRが30mL/min/1.73m2未満(Schwartz式を用いて算出) 4.1歳未満の場合は、クレアチニンが以下の閾値以上 実年齢:28日齢以上2ヵ月齢未満 クレアチニン値(mg/dL):0.6*以上 実年齢:2ヵ月齢以上1歳未満 クレアチニン値(mg/dL):0.5*以上 *クレアチニン値が97.5パーセンタイル又は年齢に対する上限以上 5.スクリーニング時の妊娠検査で陽性(妊娠可能な女性のみ) 6.腎代替療法による治療中(間欠的血液透析、腹膜透析、持続的腎代替療法) |
副作用(全グレード)が認められた被験者の割合は15.1%(8/53例)であった。5%以上でみられた主な副作用は、ALT増加5.7%(3/53例)であった。
注3)7点順序尺度[スコア1:死亡、2:入院かつECMO又は侵襲的人工呼吸器による管理、3:入院かつ非侵襲的換気又は高流量酸素による管理、4:入院かつ低流量酸素による管理、5:入院しており、酸素吸入を要しないがSARS-CoV-2による感染症に関わらず継続的な治療を要する、6:入院しており、酸素吸入及び継続的な治療は要しない(ただし、プロトコルに従った本剤の投与は除く)、7:退院]
注4)ベースライン時の臨床状態スコア(7点順序尺度)が2〜5から6又は7に改善した場合、又はベースライン時のスコアが6から7に改善した場合
18. 薬効薬理
18.1 作用機序
レムデシビルはアデノシンヌクレオシド類似体のプロドラッグである。レムデシビルは、細胞内に分布し、加水分解による代謝を経て、最終的にリン酸化されて薬理学的に活性を有するヌクレオシド三リン酸型の活性代謝物を生成する。活性代謝物はアデノシン三リン酸(ATP)の類似体として、SARS-CoV-2 RNA依存性RNAポリメラーゼによって新たに合成されるRNA鎖に天然基質ATPと競合して取り込まれ、ウイルスの複製におけるRNA鎖の伸長反応を取り込みから少し遅れて停止させる。活性代謝物は、ヒト由来のDNAポリメラーゼα、β及びRNAポリメラーゼII、並びにミトコンドリアDNAポリメラーゼγ及びミトコンドリアRNAポリメラーゼに対する阻害作用(IC50値)はいずれも>200μMであった。
18.2 In vitro抗ウイルス活性
レムデシビルは、SARS-CoV-2の臨床分離株に対して、薬剤添加48時間後におけるヒト初代培養気道上皮細胞での50%有効濃度(EC50)は9.9nMであった。また、継代培養ヒト肺上皮細胞株Calu-3及びA549-hACE2でSARS-CoV-2の複製を阻害し、EC50は薬剤添加72時間後及び48時間後でそれぞれ280nM及び115nMであった8)9)。なお、ウイルスRNA依存性RNAポリメラーゼを構成するNsp12のアミノ酸置換P323Lを含むSARS-CoV-2変異体の臨床分離株(alpha株(B.1.1.7系統)、beta株(B.1.351系統)、gamma株(P.1系統)、delta株(B.1.617.2系統)、epsilon株(B.1.429系統)、kappa株(B.1.617.1系統)、lambda株(C.37系統)、iota株(B.1.526系統)、zeta株(P.2系統)及びomicron株(B.1.1.529/BA.1、BA.2、BA.2.12.1、BA.2.75、BA.4、BA.4.6、BA.5、BF.5、BF.7、BQ.1、BQ.1.1、CH.1.1、XBB及びXBB.1.5系統))に対するNタンパク質ELISAアッセイでは、これら臨床分離株のEC50は初期のSARS-CoV-2の系統(A系統)と比較して0.15〜2.3倍であった(A549-ACE2-TMPRSS2細胞株)10)。
18.3 薬剤耐性
18.3.1 In vitro試験
培養細胞系では、レムデシビルに対する感受性が低下したSARS-CoV-2分離株が確認された。GS-441524(レムデシビルの代謝物であるヌクレオシド類似体)を用いた耐性発現試験において、レムデシビルに対する耐性変異としてNsp12のアミノ酸置換V166A、N198S、S759A、V792I、C799F及びC799Rが同定された。各置換を導入した組換えSARS-CoV-2では、レムデシビルに対して1.7〜3.5倍の感受性低下を示した11)。Nsp12のアミノ酸置換P323Lを有するSARS-CoV-2分離株を用いたレムデシビルによる耐性発現試験では、Nsp12のアミノ酸置換V166Lが同定された。P323L単独又はP323L+V166L重複置換を導入した組換えSARS-CoV-2では、レムデシビルに対してそれぞれ1.3倍及び1.5倍の感受性変化を示した12)。げっ歯類CoVのマウス肝炎ウイルスを用いたレムデシビルのin vitro耐性解析では、RNA依存性RNAポリメラーゼで全てのCoVに保存された残基において、2カ所の変異(F476L及びV553L)が確認され、レムデシビルに対して5.6倍の感受性低下を示した。この変異体はin vitroでウイルス複製能が低下した。同様の変異(F480L及びV557L)をSARS-CoVに導入したとき、培養細胞内でレムデシビルに対して6倍の感受性低下を示し、SARS-CoV感染マウスモデルにおいてウイルスの病原性が減弱した。また、Nsp12にF480L及びV557Lの各変異を導入した組換えSARS-CoV-2では、レムデシビルに対して2倍の感受性低下を示した11)。
18.3.2 臨床試験
NIAID ACTT-1試験では、ベースライン及びベースライン後におけるSARS-CoV-2のRNA依存性RNAポリメラーゼの塩基配列データが得られた本剤群31例のうち、12例で本剤投与後にアミノ酸置換が認められた。本剤群の12例で認められたアミノ酸置換は24種類であり、このうちV792I及びC799F(各1例)はin vitro耐性発現試験で既にレムデシビルに対する耐性変異として特定されており、それぞれ2.2-3.2倍及び2.5-3.5倍の感受性低下を示した11)16)。
GS-US-540-9012試験では、ベースライン及びベースライン後におけるSARS-CoV-2のRNA依存性RNAポリメラーゼの塩基配列データが得られた本剤群115例のうち、8例で本剤投与後にアミノ酸置換が認められた。本剤群の8例で認められたアミノ酸置換は7種類であり、このうちA376V(1例)は、レプリコンアッセイにおいて12.6倍の感受性低下を示した17)。
18.4 動物モデルにおける治療効果
SARS-CoV-2接種12時間後のアカゲザルSARS-CoV-2感染モデルに、投与初日はレムデシビル10mg/kgで1日1回、その後は5mg/kgで1日1回を静脈内ボーラス投与したところ、溶媒対照と比較して、呼吸器系疾患の臨床徴候が改善し、肺病理像及び肺病変所見並びに肺ウイルスRNA量が減少した。
19. 有効成分に関する理化学的知見
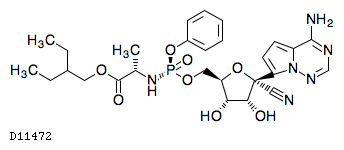
19.1. レムデシビル
| 一般的名称 | レムデシビル |
|---|---|
| 一般的名称(欧名) | Remdesivir |
| 化学名 | 2-Ethylbutyl N-{(S)-[2-C-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-2,5-anhydro-D-altrononitril-6-O-yl]phenoxyphosphoryl}-L-alaninate |
| 分子式 | C27H35N6O8P |
| 分子量 | 602.58 |
| 融点 | 138℃ |
| 物理化学的性状 | 白色〜微黄白色又は黄色の固体 |
| 溶解性 | メタノール、テトラヒドロフランに溶けやすく、エタノールにやや溶けやすく、酢酸イソプロピルに溶けにくい。 |
| 分配係数 | log P=3.2 |
| KEGG DRUG |
|
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
1バイアル
23. 主要文献
- FACT SHEET FOR HEALTH CARE PROVIDERS EMERGENCY USE AUTHORIZATION(EUA)OF REMDESIVIR(GS-5734)
- 社内資料(レムデシビル治験薬概要書)
- 社内資料(NIAID ACTT-1試験)
- 社内資料(GS-US-540-5773試験)
- Goldman JD,et al., N Engl J Med., 383 (19), 1827-1837, (2020) »PubMed
- 社内資料(GS-US-540-5774試験)
- Spinner CD,et al., JAMA, 324 (11), 1048-1057, (2020) »PubMed
- Pruijssers AJ,et al., Cell Rep., 32 (3), 107940, (2020) »PubMed
- Xie X,et al., Nat Commun., 11 (1), 5214, (2020) »PubMed
- 社内資料(PC-540-2026試験、PC-540-2034試験、PC-540-2038試験、PC-540-2039試験、PC-540-2044試験、PC-540-2046試験)
- 社内資料(PC-540-2028試験)
- 社内資料(PC-540-2029試験)
- Gottlieb RL,et al., N Engl J Med., (2021), (Published online December 22,2021.doi:10.1056/NEJMoa2116846) »PubMed
- 社内資料(GS-US-540-9013試験)
- 社内資料(REP-22251 In-Use安定性試験)
- 社内資料(PC-540-2033試験)
- 社内資料(PC-540-2040試験)
- 社内資料(GS-US-540-5823試験)
- 社内資料(IMPAACT 2032[CO-US-540-5961試験])
- Bertrand K et al., Concentrations of remdesivir and its metabolite GS-441524 in human milk from lactating individuals diagnosed with COVID-19,Abstract,APHA Annual Meeting & Expo 2022, (2022)
- Wada YS,et al., J Hum Lact., 38 (2), 248-251, (2022) »PubMed
- 社内資料(GS-US-540-9014試験)
- 社内資料(GS-US-540-5912試験、GS-US-540-9015試験、CTRA-2022-1068、QP-2023-1074)
24. 文献請求先及び問い合わせ先
文献請求先
ギリアド・サイエンシズ株式会社
メディカルサポートセンター 受付時間:9:00〜17:30(土・日・祝日及び会社休日を除く)
〒100-6616
東京都千代田区丸の内一丁目9番2号 グラントウキョウサウスタワー
電話:フリーダイアル 0120-506-295
FAX:03-5958-2959
製品情報問い合わせ先
ギリアド・サイエンシズ株式会社
メディカルサポートセンター 受付時間:9:00〜17:30(土・日・祝日及び会社休日を除く)
〒100-6616
東京都千代田区丸の内一丁目9番2号 グラントウキョウサウスタワー
電話:フリーダイアル 0120-506-295
FAX:03-5958-2959
26. 製造販売業者等
26.1 製造販売元
ギリアド・サイエンシズ株式会社
東京都千代田区丸の内1-9-2 グラントウキョウサウスタワー