医薬品情報
| 総称名 | ベリキューボ |
|---|---|
| 一般名 | ベルイシグアト |
| 欧文一般名 | Vericiguat |
| 製剤名 | ベルイシグアト錠 |
| 薬効分類名 | 慢性心不全治療剤 可溶性グアニル酸シクラーゼ(sGC)刺激剤 |
| 薬効分類番号 | 2190 |
| ATCコード | C01DX22 |
| KEGG DRUG |
D11051
ベルイシグアト
|
| JAPIC | 添付文書(PDF) |
添付文書情報2022年9月 改訂(第3版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ベリキューボ錠2.5mg | Verquvo tablets 2.5mg | バイエル薬品 | 2190042F1021 | 124.5円/錠 | 処方箋医薬品注) |
| ベリキューボ錠5mg | Verquvo tablets 5mg | バイエル薬品 | 2190042F2028 | 217.9円/錠 | 処方箋医薬品注) |
| ベリキューボ錠10mg | Verquvo tablets 10mg | バイエル薬品 | 2190042F3024 | 380.9円/錠 | 処方箋医薬品注) |
2. 禁忌
次の患者には投与しないこと
2.1 本剤の成分に対し過敏症の既往歴のある患者
2.2 可溶性グアニル酸シクラーゼ(sGC)刺激薬(リオシグアト)を投与中の患者[10.1参照]
4. 効能または効果
慢性心不全
ただし、慢性心不全の標準的な治療を受けている患者に限る。
5. 効能または効果に関連する注意
5.1 左室駆出率の保たれた慢性心不全における本剤の有効性及び安全性は確立していないため、左室駆出率の低下した慢性心不全患者に投与すること。
6. 用法及び用量
通常、成人にはベルイシグアトとして、1回2.5mgを1日1回食後経口投与から開始し、2週間間隔で1回投与量を5mg及び10mgに段階的に増量する。なお、血圧等患者の状態に応じて適宜減量する。
7. 用法及び用量に関連する注意
定期的に血圧測定を行い、臨床試験で用いられた以下の基準を参考に本剤の用量を調節すること。[8.1、9.1.1、11.1.1、17.1.1参照]
| 収縮期血圧(mmHg)・低血圧症状 | 1回投与量の調節 |
| 収縮期血圧が100mmHg以上 | ・2.5又は5mgの場合:1段階増量する。 ・10mgの場合:用量を維持する。 |
| 収縮期血圧が90mmHg以上100mmHg未満 | 用量を維持する。 |
| 収縮期血圧が90mmHg未満で低血圧症状を示さない場合 | ・2.5mgの場合:投与を中断する。 ・5又は10mgの場合:1段階減量する。 |
| 収縮期血圧が90mmHg未満で低血圧症状がある場合 | 投与を中断する。 |
8. 重要な基本的注意
8.1 本剤は血管を拡張し血圧を低下させる作用を有しており、症候性低血圧があらわれるおそれがある。血液量減少、重度の左室流出路閉塞、安静時低血圧、自律神経機能障害、低血圧の既往のある患者や、降圧剤、利尿剤、硝酸剤等の降圧作用を有する薬剤を投与中の患者では、血圧等患者の状態を十分に観察しながら慎重に投与すること。[7.、9.1.1、10.2、11.1.1参照]
8.2 めまいがあらわれることがあるので、高所作業、自動車の運転等危険を伴う機械を操作する際には注意させること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 投与前の収縮期血圧が100mmHg未満又は症候性低血圧の患者
9.2 腎機能障害患者
9.2.1 eGFR15mL/min/1.73m2未満の腎機能障害患者又は透析中の患者
9.3 肝機能障害患者
9.3.1 重度の肝機能障害患者(Child-Pugh分類C)
9.4 生殖能を有する者
妊娠する可能性のある女性は本剤の投与中及び投与終了後一定期間は確実な避妊法を用いること。[9.5参照]
9.5 妊婦
妊婦又は妊娠している可能性のある女性には投与しないことが望ましい。ウサギにおける流産及び全胚吸収がヒトの6倍以上の全身曝露量で、ラットにおける出生児の死亡率の増加及び体重増加抑制がそれぞれヒトの49倍及び21倍以上の全身曝露量で発現することが報告されている。[9.4参照]
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。ヒトにおける乳汁中移行性については不明である。動物実験(ラット、静脈内投与)で乳汁中に移行することが報告されている。
9.7 小児等
小児等を対象とした臨床試験は実施していない。
10. 相互作用
10.1 併用禁忌
| 可溶性グアニル酸シクラーゼ(sGC)刺激薬 リオシグアト(アデムパス) [2.2参照] | 症候性低血圧を起こすおそれがある。 | 細胞内cGMP濃度が増加し、降圧作用を増強するおそれがある。 |
10.2 併用注意
11. 副作用
11.1 重大な副作用
11.2 その他の副作用
| 1〜10%未満 | 1%未満 | 頻度不明 | |
| 血液およびリンパ系障害 | 貧血 | ||
| 神経系障害 | 浮動性めまい | 頭痛 | |
| 胃腸障害 | 消化不良、胃食道逆流性疾患、悪心、嘔吐 |
13. 過量投与
13.1 症状
過度の血圧低下等が起こる可能性がある。
13.2 処置
本剤はタンパク結合率が高いので、血液透析による除去は期待できない。
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
健康成人男性36例を対象として本剤1.25、5、7.5又は10mgを空腹時単回経口投与したとき、血漿中未変化体濃度は投与1.0〜2.5時間後に最高血漿中濃度に達した後、17.9〜22.3時間の半減期で低下した。
| 1.25mg (n=9) | 5mg (n=9) | 7.5mg (n=9) | 10mg (n=9) | |
| AUC(μg・h/L) | 1190/17.5 | 4140/21.1 | 5140/38.6 | 7410/30.5 |
| Cmax(μg/L) | 61.8/28.5 | 236/28.9 | 324/39.6 | 365/37.1 |
| tmax※(h) | 1.0 (0.75-3.0) | 1.0 (0.75-4.0) | 1.0 (0.5-2.5) | 2.5 (0.75-4.0) |
| t1/2(h) | 22.3/18.2 | 21.1/31.0 | 17.9/17.9 | 18.3/20.1 |
16.1.2 反復投与
健康成人男性35例を対象として本剤1.25、5、7.5又は10mgを1日1回7日間空腹時反復経口投与したとき、単回投与時と比較して本剤の薬物動態特性に大きな変化はなく、時間依存性はみられなかった。最終投与時のAUC(0-24)は単回投与時の1.4〜1.7倍であった。
| 1.25mg (n=9) | 5mg (n=8) | 7.5mg (n=9) | 10mg (n=9) | |
| AUC(0-24)ss(μg・h/L) | 1170/14.5 | 3670/23.4 | 4810/27.6 | 6170/29.9 |
| Cmax,ss(μg/L) | 89.2/18.6 | 289/25.1 | 407/24.2 | 472/30.6 |
| tmax※(h) | 1.0 (0.75-2.5) | 1.75 (0.75-4.0) | 2.5 (0.75-2.5) | 2.5 (0.75-2.5) |
| t1/2(h) | 27.0/23.6 | 23.5/30.6 | 22.1/15.9 | 20.7/25.2 |
16.2 吸収
16.2.1 バイオアベイラビリティ
本剤の食後投与したときの絶対的バイオアベイラビリティは93%であった(外国人データ)。
16.2.2 食事の影響
本剤を高脂肪・高カロリー食摂取後に単回経口投与したところ、tmax(中央値)は空腹時投与時の投与後1時間から4時間に延長し、薬物動態パラメータの個体間変動の程度は減少した。空腹時投与時と比較して、本剤5mg(5mg錠1錠)食後投与時のAUCは19%、Cmaxは9%上昇し、本剤10mg(10mg錠1錠)食後投与時のAUCは44%、Cmaxは41%上昇した。低脂肪・高炭水化物食摂取後投与時にも同様の食事の影響がみられた(外国人データ)。
16.3 分布
16.3.1 分布容積
本剤の静脈内投与したときの定常状態における分布容積は約44Lであった(外国人データ)。
16.3.2 タンパク結合率
本剤のヒト血漿タンパク結合率は約98%で、主にアルブミンと結合した(in vitro)。
16.4 代謝
本剤の主代謝経路はN-グルクロン酸抱合体(代謝物M-1)へのグルクロン酸抱合である。主代謝酵素はUGT1A9及びUGT1A1である。CYPによる酸化代謝は本剤の主代謝経路ではない(in vitro、in vivo)。
16.5 排泄
健康成人男性10例を対象として本剤を静脈内投与したときの全身クリアランスは約1.6L/hであった。健康成人男性6例を対象として[14C]ベルイシグアトを空腹時単回経口投与したとき、投与された総放射能の53%が尿中に、45%が糞中に排泄された。また、未変化体は投与された総放射能の9%が尿中に、43%が糞中に排泄され、代謝物M-1は投与された総放射能の41%が尿中に排泄され、糞中には排泄されなかった(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
中等度(eGFR30mL/min/1.73m2以上60mL/min/1.73m2未満、14例)又は重度(eGFR30mL/min/1.73m2未満、8例)の腎機能障害患者に本剤2.5mgを食後に単回経口投与したとき、本剤の非結合型AUCは健康成人(8例)と比較してそれぞれ76%及び128%高かった(外国人データ)。[9.2.1参照]
16.6.2 肝機能障害患者
軽度(Child-Pugh分類A、9例)及び中等度(Child-Pugh分類B、9例)の肝機能障害を有する患者における本剤の非結合型AUCは健康成人(9例)と比較して8%及び41%高かった(外国人データ)。[9.3.1参照]
16.7 薬物相互作用
16.7.1 オメプラゾール/制酸剤
健康成人10例を対象としてオメプラゾール40mgを1日1回4日間前投与した翌日に本剤5mgを空腹時注)併用投与したとき、本剤5mgを単独投与したときと比較して、本剤のAUC及びCmaxは32%及び50%低下した。水酸化アルミニウム・水酸化マグネシウム懸濁剤10mLと本剤5mgを空腹時注)併用投与したときには、本剤単独投与時と比較して、本剤のAUC及びCmaxは27%及び46%低下した(外国人データ)。[16.2.1参照]
16.7.2 シルデナフィル
16.7.3 サクビトリルバルサルタンナトリウム水和物
健康成人15例を対象としてサクビトリル/バルサルタン97/103mgを1日2回反復投与した定常状態下において本剤2.5mgを単回併用投与したとき、本剤2.5mgを単独単回投与したときと比較して、本剤のAUC(0-24)及びCmaxは7%及び9%低下した。サクビトリル/バルサルタン97/103mgを1日2回単独反復投与したときと比較して、本剤2.5mg(1日1回)と反復併用投与したときには、バルサルタンのAUC(0-12)及びCmaxは12%及び13%、サクビトリルのAUC(0-12)及びCmaxは8%及び18%、サクビトリルの活性代謝物LBQ657のAUC(0-12)及びCmaxは1%及び2%上昇した。座位血圧に本剤併用投与による影響は認められなかった(外国人データ)。
16.7.4 硝酸剤
16.7.5 ジゴキシン
健康成人25例を対象として本剤10mgとジゴキシン0.375mgを1日1回反復併用投与したときの定常状態におけるジゴキシンのAUCτ,mdは、ジゴキシン0.375mgを単独で1日1回反復投与したときと比較して4%上昇した。ジゴキシン0.375mgを1日1回反復投与したときの定常状態下において本剤10mgを単回投与したとき、本剤10mgを単独で単回投与したときと比較して、本剤のAUCは2%低下しCmaxは1%上昇した(外国人データ)。
16.7.6 アスピリン
健康成人14例を対象としてアスピリン500mgを投与した翌日に本剤15mg注)を併用投与したときの本剤のAUC及びCmaxは、本剤15mg注)を単独投与したときと比較して、それぞれ5%及び7%低下した。本剤は出血時間及び抗凝固作用に影響せず、アスピリン投与時と本剤併用投与時の出血時間及び抗凝固作用にも変化はみられなかった(外国人データ)。
16.7.7 ワルファリン
健康成人23例を対象として本剤10mgを1日1回反復投与した定常状態においてワルファリン25mgを併用投与したときのPT、第VII、第II及び第X因子のAUC(0-96)は、ワルファリン25mgをプラセボと併用投与したときの97.4〜100.4%であった。薬物動態にも相互作用はみられなかった(外国人データ)。
注)本剤の承認用法及び用量は、「1回2.5mgを1日1回食後経口投与から開始し、2週間間隔で1回投与量を5mg及び10mgに段階的に増量する。なお、血圧等患者の状態に応じて適宜減量する。」である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国際共同第III相試験
ACE阻害薬、ARB、β遮断薬及びMRA等の慢性心不全の標準治療を受けている左室駆出率(LVEF)の低下した慢性心不全患者※1(LVEF45%未満、NYHA心機能分類II〜IV度)5050例(日本人319例を含む)を対象としたプラセボ対照無作為化二重盲検並行群間比較試験を実施した。プラセボ又は本剤を1日1回食後に経口投与し、本剤は2.5mgより投与を開始し、投与2週間後に5mg、投与4週間後に目標用量である10mgに増量した。なお、血圧、忍容性等に応じて、2週間程度の間隔で1日1回2.5〜10mgの範囲で用量を調節した。有効性主要評価項目(心血管死又は心不全による初回入院の複合エンドポイント)の評価期間の中央値は11ヵ月であり、投与期間は平均1年、最長2.6年であった。
※1:無作為割付け前6ヵ月以内に心不全による入院の既往又は無作為割付け前3ヵ月以内に入院を要しない静注用利尿薬の投与経験がある患者が組み入れられた。
有効性主要評価項目である心血管死又は心不全による初回入院の複合エンドポイント、並びに副次評価項目である心血管死及び心不全による初回入院の発現割合及び発現率は下表のとおりであった。
| 本剤群 n=2526 | プラセボ群 n=2524 | ハザード比 (95%信頼区間)※2 | P値※3 | ||
| n(%) | 発現率※1 | n(%) | 発現率※1 | ||
| 有効性主要評価項目 (心血管死又は心不全による初回入院の複合エンドポイント) | |||||
| 897 (35.5) | 33.6 | 972 (38.5) | 37.8 | 0.90 (0.82-0.98) | 0.019 |
| 副次評価項目(心血管死) | |||||
| 414 (16.4) | 12.9 | 441 (17.5) | 13.9 | 0.93 (0.81-1.06) | 0.269 |
| 副次評価項目(心不全による初回入院) | |||||
| 691 (27.4) | 25.9 | 747 (29.6) | 29.1 | 0.90 (0.81-1.00) | 0.048 |
日本人集団における有効性主要評価項目である心血管死又は心不全による初回入院の複合エンドポイント、並びに副次評価項目である心血管死及び心不全による初回入院の発現割合及び発現率は下表のとおりであった。
| 本剤群 n=161 | プラセボ群 n=158 | ハザード比 (95%信頼区間)※2 | ||
| n(%) | 発現率※1 | n(%) | 発現率※1 | |
| 有効性主要評価項目 (心血管死又は心不全による初回入院の複合エンドポイント) | ||||
| 49 (30.4) | 27.6 | 49 (31.0) | 30.3 | 0.93 (0.63-1.39) |
| 副次評価項目(心血管死) | ||||
| 23 (14.3) | 11.0 | 11 (7.0) | 5.4 | 2.01 (0.98-4.12) |
| 副次評価項目(心不全による初回入院) | ||||
| 38 (23.6) | 21.4 | 44 (27.8) | 27.2 | 0.82 (0.53-1.26) |
副作用は2519例中367例(14.6%)に認められ、主な副作用は、低血圧172例(6.8%)、浮動性めまい37例(1.5%)、悪心19例(0.8%)等であった。
日本人集団における副作用は161例中13例(8.1%)に認められ、主な副作用は低血圧3例(1.9%)であった。
なお、症候性低血圧(有害事象)及び失神(有害事象)は本剤群で9.1%及び4.0%、プラセボ群で7.9%及び3.5%であった。また、日本人集団における症候性低血圧(有害事象)及び失神(有害事象)は本剤群で5.0%及び3.1%、プラセボ群で3.8%及び1.9%であった。
日本人集団における副作用は161例中13例(8.1%)に認められ、主な副作用は低血圧3例(1.9%)であった。
なお、症候性低血圧(有害事象)及び失神(有害事象)は本剤群で9.1%及び4.0%、プラセボ群で7.9%及び3.5%であった。また、日本人集団における症候性低血圧(有害事象)及び失神(有害事象)は本剤群で5.0%及び3.1%、プラセボ群で3.8%及び1.9%であった。
17.3 その他
冠動脈疾患患者74例を対象としてプラセボ対照二重盲検法により本剤2.5、5、10mg及びプラセボを1日1回14日間反復投与した。本剤10mg反復投与後におけるQTcF間隔のベースラインからの最大変化量(プラセボとの差)は平均6msec未満で、両側90%信頼区間の上限値は10msec未満であった(外国人データ)。
18. 薬効薬理
18.1 作用機序
本剤は、心筋収縮力、血管緊張、心臓リモデリング等の生理機能の調節に関わる環状グアノシン一リン酸(cGMP)を生成する可溶性グアニル酸シクラーゼ(sGC)を、一酸化窒素(NO)非依存的に、またNOを介して刺激し、cGMPの生成を促進する。
18.2 血行動態に及ぼす作用
本剤は、麻酔下のラットにおいて、平均動脈圧を低下させ、心拍数を反射性に増加させた。また、麻酔下のイヌにおいて、平均動脈圧、肺動脈圧及び全身血管抵抗を低下させ、心拍数を反射性に増加させた。
18.3 心不全モデルにおける作用
19. 有効成分に関する理化学的知見
19.1. ベルイシグアト
| 一般的名称 | ベルイシグアト |
|---|---|
| 一般的名称(欧名) | Vericiguat |
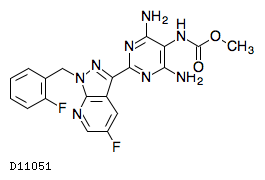
| 化学名 | Methyl(4,6-diamino-2-{5-fluoro-1-[(2-fluorophenyl)methyl]-1H-pyrazolo[3,4-b]pyridin-3-yl}pyrimidin-5-yl)carbamate |
| 分子式 | C19H16F2N8O2 |
| 分子量 | 426.38 |
| 物理化学的性状 | 本品は白色〜帯黄色の粉末である。 |
| KEGG DRUG |
|
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
<ベリキューボ錠2.5mg>
100錠[10錠(PTP)×10]
<ベリキューボ錠5mg>
100錠[10錠(PTP)×10]
<ベリキューボ錠10mg>
100錠[10錠(PTP)×10]
23. 主要文献
24. 文献請求先及び問い合わせ先
文献請求先
バイエル薬品株式会社
メディカルインフォメーション
〒530-0001
大阪市北区梅田二丁目4番9号
文献請求先
製品情報問い合わせ先
バイエル薬品株式会社
電話:0120-106-398
バイエル医療用医薬品のお問い合わせ先
26. 製造販売業者等
26.1 製造販売元
バイエル薬品株式会社
大阪市北区梅田二丁目4番9号