医薬品情報
| 総称名 | レベチラセタム |
|---|---|
| 一般名 | レベチラセタム |
| 欧文一般名 | Levetiracetam |
| 製剤名 | レベチラセタムドライシロップ |
| 薬効分類名 | 抗てんかん剤 |
| 薬効分類番号 | 1139 |
| ATCコード | N03AX14 |
| KEGG DRUG |
D00709
レベチラセタム
|
| JAPIC | 添付文書(PDF) |
添付文書情報2026年3月 改訂(第4版)

|
2.禁忌 4.効能または効果 6.用法及び用量 7.用法及び用量に関連する注意 8.重要な基本的注意 9.特定の背景を有する患者に関する注意 11.副作用 13.過量投与 15.その他の注意 16.薬物動態 17.臨床成績 18.薬効薬理 19.有効成分に関する理化学的知見 22.包装 23.主要文献 24.文献請求先及び問い合わせ先 26.製造販売業者等 |
商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| レベチラセタムDS50%「サワイ」 (後発品) | LEVETIRACETAM DS[SAWAI] | 沢井製薬 | 1139010R1063 | 57.9円/g | 処方箋医薬品注) |
2. 禁忌
次の患者には投与しないこと
本剤の成分又はピロリドン誘導体に対し過敏症の既往歴のある患者
4. 効能または効果
○てんかん患者の部分発作(二次性全般化発作を含む)
○他の抗てんかん薬で十分な効果が認められないてんかん患者の強直間代発作に対する抗てんかん薬との併用療法
6. 用法及び用量
成人
通常、成人にはレベチラセタムとして1日1000mg(ドライシロップとして2g)を1日2回に分けて用時溶解して経口投与する。なお、症状により1日3000mg(ドライシロップとして6g)を超えない範囲で適宜増減するが、増量は2週間以上の間隔をあけて1日用量として1000mg(ドライシロップとして2g)以下ずつ行うこと。
小児
通常、4歳以上の小児にはレベチラセタムとして1日20mg/kg(ドライシロップとして40mg/kg)を1日2回に分けて用時溶解して経口投与する。なお、症状により1日60mg/kg(ドライシロップとして120mg/kg)を超えない範囲で適宜増減するが、増量は2週間以上の間隔をあけて1日用量として20mg/kg(ドライシロップとして40mg/kg)以下ずつ行うこと。ただし、体重50kg以上の小児では、成人と同じ用法・用量を用いること。
7. 用法及び用量に関連する注意
7.1 本剤を強直間代発作に対して使用する場合には、他の抗てんかん薬と併用すること。強直間代発作に対する本剤単独投与での臨床試験は実施していない。
7.2 腎機能障害を有する成人患者に本剤を投与する場合は、下表に示すクレアチニンクリアランス値を参考として本剤の投与量及び投与間隔を調節すること。また、血液透析を受けている成人患者では、クレアチニンクリアランス値に応じた1日用量に加えて、血液透析を実施した後に本剤の追加投与を行うこと。なお、ここで示している用法及び用量はシミュレーション結果に基づくものであることから、患者ごとに慎重に観察しながら、用法及び用量を調節すること。また、腎機能障害を有する小児患者についても腎機能の程度に応じて、本剤の投与量及び投与間隔を調節するなど慎重に投与すること。[9.2.1、9.2.2、9.8、16.6.1、16.6.2参照]
| クレアチニンクリアランス(mL/min) | ≧80 | ≧50-<80 | ≧30-<50 | <30 | 透析中の腎不全患者 | 血液透析後の補充用量 |
| 1日投与量 | 1000〜3000mg | 1000〜2000mg | 500〜1500mg | 500〜1000mg | 500〜1000mg | / |
| 通常投与量 | 1回 500mg 1日2回 | 1回 500mg 1日2回 | 1回 250mg 1日2回 | 1回 250mg 1日2回 | 1回 500mg 1日1回 | 250mg |
| 最高投与量 | 1回 1500mg 1日2回 | 1回 1000mg 1日2回 | 1回 750mg 1日2回 | 1回 500mg 1日2回 | 1回 1000mg 1日1回 | 500mg |
8. 重要な基本的注意
8.1 連用中における投与量の急激な減量ないし投与中止により、てんかん発作の増悪又はてんかん重積状態があらわれることがあるので、投与を中止する場合には、少なくとも2週間以上かけて徐々に減量するなど慎重に行うこと。
8.2 眠気、注意力・集中力・反射運動能力等の低下が起こることがある。自動車の運転等危険を伴う機械操作の適否は、関連学会の留意事項1)を十分理解の上、医師が慎重に判断し、危険を伴う機械操作を行う場合には十分な注意が必要であることを適切に患者に指導すること。また、眠気等があらわれた場合には、自動車の運転等危険を伴う機械の操作に従事しないよう、患者に指導すること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 フェニルケトン尿症の患者
本剤は1g中30mgのアスパルテーム(L-フェニルアラニン化合物)を含有するため、フェニルケトン尿症の症状を増悪させるおそれがある。
9.2 腎機能障害患者
9.3 肝機能障害患者
9.5 妊婦
9.5.1 妊婦又は妊娠している可能性のある女性には、以下のようなリスクを考慮し治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。
・ヒトにおいて、妊娠中にレベチラセタムの血中濃度が低下したとの報告があり、第3トリメスター期間に多く、最大で妊娠前の60%となったとの報告がある。
・ラットにおいて胎児移行性が認められている。
・動物実験において、ラットではヒトへの曝露量と同程度以上の曝露で骨格変異及び軽度の骨格異常の増加、成長遅延、児の死亡率増加が認められ、ウサギでは、ヒトへの曝露量の4〜5倍の曝露で胚致死、骨格異常の増加及び奇形の増加が認められている。
9.5.2 本剤を投与した妊婦から出生した児において、新生児薬物離脱症候群があらわれることがある。
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。
ヒト乳汁中へ移行することが報告されている。
ヒト乳汁中へ移行することが報告されている。
9.7 小児等
低出生体重児又は新生児を対象とした臨床試験は国内・海外ともに実施していない。
9.8 高齢者
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 中毒性表皮壊死融解症(Toxic Epidermal Necrolysis:TEN)、皮膚粘膜眼症候群(Stevens-Johnson症候群)(いずれも頻度不明)
発熱、紅斑、水疱・びらん、そう痒、咽頭痛、眼充血、口内炎等の異常が認められた場合には投与を中止し、適切な処置を行うこと。
11.1.2 薬剤性過敏症症候群(頻度不明)
初期症状として発疹、発熱がみられ、更に肝機能障害、リンパ節腫脹、白血球増加、好酸球増多、異型リンパ球出現等を伴う遅発性の重篤な過敏症状があらわれることがある。なお、ヒトヘルペスウイルス6(HHV-6)等のウイルスの再活性化を伴うことが多く、投与中止後も発疹、発熱、肝機能障害等の症状が再燃あるいは遷延化することがあるので注意すること2)。
11.1.3 重篤な血液障害(頻度不明)
汎血球減少、無顆粒球症、白血球減少、好中球減少、血小板減少があらわれることがある。
11.1.4 肝不全、肝炎(いずれも頻度不明)
肝不全、肝炎等の重篤な肝障害があらわれることがある。
11.1.5 膵炎(頻度不明)
激しい腹痛、発熱、嘔気、嘔吐等の症状があらわれたり、膵酵素値の上昇が認められた場合には投与を中止し、適切な処置を行うこと。
11.1.6 攻撃性、自殺企図(いずれも1%未満)
11.1.7 横紋筋融解症(頻度不明)
筋肉痛、脱力感、CK上昇、血中及び尿中ミオグロビン上昇等があらわれた場合には投与を中止し、適切な処置を行うこと。
11.1.8 急性腎障害(頻度不明)
11.1.9 悪性症候群(頻度不明)
発熱、筋強剛、血清CK上昇、頻脈、血圧の変動、意識障害、発汗過多、白血球の増加等があらわれた場合には投与を中止し、体冷却、水分補給、呼吸管理等の適切な処置を行うこと。また、ミオグロビン尿を伴う腎機能の低下がみられることがある。
11.2 その他の副作用
| 3%以上 | 1〜3%未満 | 1%未満 | 頻度不明 | |
| 精神神経系 | 浮動性めまい(10.4%)、頭痛(11.8%)、不眠症、傾眠(27.9%) | 感覚鈍麻、気分変動、振戦、易刺激性、痙攣、抑うつ | 激越、健忘、注意力障害、幻覚、運動過多、記憶障害、錯感覚、思考異常、平衡障害、感情不安定、異常行動、協調運動異常、怒り、ジスキネジー、不安、体位性めまい、睡眠障害、緊張性頭痛、精神病性障害、パニック発作、譫妄 | 錯乱状態、敵意、気分動揺、神経過敏、人格障害、精神運動亢進、舞踏アテトーゼ運動、嗜眠、てんかん増悪、強迫性障害 |
| 眼 | 複視、結膜炎 | 霧視、眼精疲労、眼そう痒症、麦粒腫 | ||
| 血液 | 好中球数減少 | 貧血、血中鉄減少、鉄欠乏性貧血、血小板数減少、白血球数増加、白血球数減少 | ||
| 循環器 | 心電図QT延長、高血圧 | |||
| 消化器 | 腹痛、便秘、下痢、胃腸炎、悪心、口内炎、嘔吐、齲歯 | 歯肉炎、痔核、胃不快感、歯痛 | 消化不良、口唇炎、歯肉腫脹、歯周炎 | |
| 肝臓 | ALP増加 | 肝機能異常 | ||
| 泌尿・生殖器 | 膀胱炎、尿中ブドウ糖陽性、尿中血陽性、尿中蛋白陽性、月経困難症 | 頻尿 | ||
| 呼吸器 | 鼻咽頭炎(30.2%)、咽喉頭疼痛、上気道の炎症 | 気管支炎、咳嗽、鼻漏、咽頭炎、インフルエンザ、鼻炎 | 鼻出血、肺炎 | |
| 代謝及び栄養 | 食欲不振 | |||
| 皮膚 | 湿疹 | 皮膚炎、そう痒症、発疹、ざ瘡 | 脱毛症、単純ヘルペス、帯状疱疹、白癬感染 | 多形紅斑、血管性浮腫 |
| 筋骨格系 | 背部痛 | 肩痛、筋肉痛、筋骨格硬直、関節痛 | 頸部痛、四肢痛、筋力低下 | |
| 感覚器 | 耳鳴 | 回転性めまい | ||
| その他 | 倦怠感、発熱、体重減少 | 血中トリグリセリド増加、胸痛、体重増加 | 無力症、疲労、末梢性浮腫、抗痙攣剤濃度増加 | 事故による外傷(皮膚裂傷等) |
13. 過量投与
13.1 症状
外国の市販後報告において、レベチラセタムを一度に15〜140g服用した例があり、傾眠、激越、攻撃性、意識レベルの低下、呼吸抑制及び昏睡が報告されている。
13.2 処置
本剤は血液透析により除去可能であり、発現している症状の程度に応じて血液透析の実施を考慮すること。[16.6.2参照]
15. その他の注意
15.1 臨床使用に基づく情報
15.1.1 海外で実施された本剤を含む複数の抗てんかん薬における、てんかん、精神疾患等を対象とした199のプラセボ対照臨床試験の検討結果において、自殺念慮及び自殺企図の発現のリスクが、抗てんかん薬の服用群でプラセボ群と比較して約2倍高く(抗てんかん薬服用群:0.43%、プラセボ群:0.24%)、抗てんかん薬の服用群では、プラセボ群と比べ1000人あたり1.9人多いと計算された(95%信頼区間:0.6-3.9)。また、てんかん患者のサブグループでは、プラセボ群と比べ1000人あたり2.4人多いと計算されている。[8.3、8.4、11.1.6参照]
15.1.2 外国人成人てんかん患者1208例を対象としたプラセボ対照臨床試験の併合解析において、非精神病性行動症状の有害事象(攻撃性、激越、怒り、不安、無力感、離人症、抑うつ、情動不安定、敵意、運動過多、易刺激性、神経過敏、神経症、人格障害)の発現率は本剤群で13.3%、プラセボ群で6.2%であった。同様に、外国人小児てんかん患者(4〜16歳)198例を対象としたプラセボ対照臨床試験における当該有害事象の発現率は本剤群で37.6%、プラセボ群で18.6%であった。
また、外国人小児てんかん患者(4〜16歳)98例を対象とした認知機能及び行動に対する影響を評価するプラセボ対照臨床試験において、探索的な検討であるが、プラセボ群と比較して攻撃的行動の悪化が示唆された。
また、外国人小児てんかん患者(4〜16歳)98例を対象とした認知機能及び行動に対する影響を評価するプラセボ対照臨床試験において、探索的な検討であるが、プラセボ群と比較して攻撃的行動の悪化が示唆された。
16. 薬物動態
16.1 血中濃度
16.1.1 成人
(1)単回投与
健康成人にレベチラセタム250、500、1000、1500、2000、3000、4000注1)、5000mg注1)(各投与量6例)を空腹時に単回経口投与したとき、すべての投与量でレベチラセタムの血漿中濃度は投与後ほぼ1時間に最高値を示し、消失半減期(t1/2)は投与量にかかわらず7〜9時間であった3)。
| 投与量(mg) | Cmax(μg/mL) | tmax(h) | AUC0-48h(μg・h/mL) | t1/2(h) |
| 250 | 6.9±1.3 | 1.0±0.6 | 56.7±6.2 | 6.9±0.9 |
| 500 | 16.4±4.8 | 1.0±0.6 | 148.7±18.4 | 7.9±1.0 |
| 1000 | 29.7±9.3 | 0.8±0.6 | 288.9±34.0 | 7.9±1.0 |
| 1500 | 40.8±7.2 | 0.8±0.3 | 458.1±50.9 | 8.1±0.4 |
| 2000 | 53.3±8.3 | 0.8±0.6 | 574.6±71.4 | 8.0±0.8 |
| 3000 | 82.9±7.4 | 0.6±0.2 | 925.2±102.1 | 7.8±0.8 |
| 4000注1) | 114.1±11.0 | 0.9±0.6 | 1248.2±152.4 | 8.6±1.0 |
| 5000注1) | 115.1±14.3 | 1.0±0.6 | 1363.3±151.9 | 8.1±0.7 |
注1)国内で承認された本剤の1日最高投与量は3000mgである。
(2)反復投与
健康成人にレベチラセタムとして1回1000mg又は1500mg(各投与量6例)を1日2回7日間投与したとき、投与1日目(初回投与時)と7日目(最終回投与時)の血漿中濃度は共に投与後約2〜3時間にCmaxを示し、その後約8時間の消失半減期で低下した。また、血漿中濃度は投与3日目には定常状態に達すると推測された4)。
| 薬物動態パラメータ | 2000mg/日(N=6) | 3000mg/日(N=6) | ||
| 初回投与時 | 最終回投与時 | 初回投与時 | 最終回投与時 | |
| Cmax(μg/mL) | 24.1±3.0 | 36.3±5.7 | 33.3±3.6 | 52.0±4.6 |
| tmax(h) | 2.2±1.2 | 2.8±1.0 | 2.2±0.8 | 2.5±1.0 |
| AUC0-12h(μg・h/mL) | 191.3±26.7 | 318.3±63.2 | 253.7±30.3 | 445.6±56.9 |
| t1/2(h) | 8.0±1.4 | 8.3±0.9 | 7.5±0.7 | 7.7±0.4 |
(3)点滴静脈内投与と経口投与の比較
健康成人25例にレベチラセタム1500mgを15分間点滴静脈内投与又は経口投与したとき、レベチラセタムの薬物動態パラメータは以下のとおりであった。経口投与時と比較して、点滴静脈内投与時のCmaxは約1.6倍高く、AUC及びt1/2は類似していた。なお、レベチラセタム経口投与時の生物学的利用率は約100%であった5)。
| 薬物動態パラメータ | 点滴静脈内投与 (N=25) | 経口投与 (N=25) | 幾何平均比注2)(90%信頼区間) |
| Cmax(μg/mL) | 97.0[27.6] | 58.9[37.0] | 1.64(1.47-1.83) |
| AUC0-t(μg・h/mL) | 472.3[15.4] | 487.4[15.9] | 0.97(0.95-0.99) |
| tmax(h) | 0.25(0.17-0.27) | 0.75(0.50-3.00) | − |
| t1/2(h) | 7.11[11.7] | 7.23[12.7] | − |
16.1.2 小児
16.1.3 母集団薬物動態解析
成人
日本人及び外国人の健康成人及びてんかん患者(クレアチニンクリアランス:49.2〜256.8mL/min)から得られた血漿中レベチラセタム濃度データを用いて、母集団薬物動態解析を行った。その結果、見かけの全身クリアランス(CL/F)に対して、体重、性別、CLCR及び併用抗てんかん薬、見かけの分布容積(V/F)に対して体重、併用抗てんかん薬及び被験者の健康状態(健康成人又はてんかん患者)が統計学的に有意な因子として推定された8)。
小児
小児(4〜16歳)及び成人(16〜55歳)のてんかん患者から得られた血漿中レベチラセタム濃度データを用いて、母集団薬物動態解析を行った。その結果、CL/Fに対して体重及び併用抗てんかん薬、V/Fに対して体重が統計学的に有意かつ臨床的に意味のある因子として推定された。小児及び成人てんかん患者の血漿中薬物濃度をシミュレーションした結果、小児てんかん患者に10〜30mg/kgを1日2回投与した際の血漿中薬物濃度は、成人てんかん患者に500〜1500mg1日2回投与した際と同様と予測された9)。
16.1.4 生物学的同等性試験
レベチラセタムDS50%「サワイ」とイーケプラドライシロップ50%を健康成人男性にそれぞれ1g(レベチラセタムとして500mg)空腹時単回経口投与(クロスオーバー法)し、血漿中レベチラセタム濃度を測定した。得られた薬物動態パラメータ(AUC、Cmax)について90%信頼区間法にて統計解析を行った結果、log(0.80)〜log(1.25)の範囲内であり、両剤の生物学的同等性が確認された10)。
| Cmax(μg/mL) | Tmax(hr) | T1/2(hr) | AUC0-36hr(μg・hr/mL) | |
| レベチラセタムDS50%「サワイ」 | 12.88±2.27 | 0.8±0.6 | 8.0±0.7 | 123.21±13.02 |
| イーケプラドライシロップ50% | 13.35±2.82 | 0.6±0.4 | 8.1±0.7 | 122.23±15.45 |
血漿中濃度ならびにAUC、Cmax等のパラメータは、被験者の選択、体液の採取回数・時間等の試験条件によって異なる可能性がある。
16.2 吸収
16.2.1 食事の影響
健康成人12例に、レベチラセタム1500mgを空腹時又は食後に単回経口投与したとき、空腹時と比べて、食後投与時ではtmaxが約1.3時間延長し、Cmaxは30%低下したが、AUCは同等であった11)。
16.3 分布
16.4 代謝
16.5 排泄
健康成人(各投与量6例)にレベチラセタム250〜5000mg注3)を空腹時に単回経口投与したとき3)、投与48時間後までの投与量に対する尿中排泄率の平均値は、未変化体として56.3〜65.3%、ucb L057として17.7〜21.9%であった。
外国人健康成人男性4例に14C-レベチラセタム500mgを単回経口投与したとき13)、投与48時間後までに投与量の92.8%の放射能が尿中から、0.1%が糞中から回収された。投与48時間後までの投与量に対する尿中排泄率は、未変化体として65.9%、ucb L057として23.7%であった。
レベチラセタムの排泄には糸球体ろ過及び尿細管再吸収が、ucb L057には糸球体ろ過と能動的尿細管分泌が関与している17)。
外国人健康成人男性4例に14C-レベチラセタム500mgを単回経口投与したとき13)、投与48時間後までに投与量の92.8%の放射能が尿中から、0.1%が糞中から回収された。投与48時間後までの投与量に対する尿中排泄率は、未変化体として65.9%、ucb L057として23.7%であった。
レベチラセタムの排泄には糸球体ろ過及び尿細管再吸収が、ucb L057には糸球体ろ過と能動的尿細管分泌が関与している17)。
注3)国内で承認された本剤の1日最高投与量は3000mgである。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
腎機能の程度の異なる成人被験者を対象に、レベチラセタムを単回経口投与したとき、見かけの全身クリアランスは腎機能正常者(CLCR:≧80mL/min/1.73m2)と比較して、軽度低下者(CLCR:50〜<80mL/min/1.73m2)では40%、中等度低下者(CLCR:30〜<50mL/min/1.73m2)で52%、重度低下者(CLCR:<30mL/min/1.73m2)で61%低下した。レベチラセタムの腎クリアランスはクレアチニンクリアランスと有意に相関した18)。[7.2、9.2.1、9.8、16.6.4参照]
| 薬物動態パラメータ | 腎機能の程度 | |||
| 正常 (N=6) | 軽度 (N=6) | 中等度 (N=6) | 重度 (N=6) | |
| CLCR(mL/min/1.73m2) | ≧80 | 50-<80 | 30-<50 | <30 |
| 投与量 | 500mg | 500mg | 250mg | 250mg |
| レベチラセタム | ||||
| Cmax(μg/mL) | 21.9[31.2] | 15.5[25.3] | 10.8[24.3] | 9.2[30.4] |
| tmax(h) | 0.5(0.5-2.0) | 1.0(0.5-2.0) | 0.5(0.5-1.0) | 0.5(0.5-1.0) |
| AUC0-t(μg・h/mL) | 166[16.5] | 248[16.9] | 169[16.5] | 212[19.1] |
| t1/2(h) | 7.6[6.9] | 12.6[11.3] | 15.5[17.5] | 19.7[26.5] |
| CL/F(mL/min/1.73m2) | 51.5[7.8] | 30.9[14.6] | 24.6[15.0] | 20.3[20.9] |
| CLR(mL/min/1.73m2) | 31.6[28.5]※ | 15.3[22.3] | 9.7[23.4] | 6.0[53.6] |
| ucb L057 | ||||
| Cmax(μg/mL) | 0.36[9.4] | 0.75[25.8] | 0.57[26.0] | 1.06[29.3] |
| tmax(h) | 5.0(2.0-8.0) | 8.0(6.0-12.0) | 12.0(8.0-12.0) | 24.0(12.0-24.0) |
| AUC0-t(μg・h/mL) | 5.9[9.7] | 22.6[45.9] | 18.7[53.4] | 57.8[57.3] |
| t1/2(h) | 12.4(11.3-15.3) | 19.0(17.3-19.9) | 20.3(19.7-23.6) | 26.8(17.2-33.3) |
16.6.2 血液透析を受けている末期腎機能障害患者
血液透析を受けている末期腎機能障害の成人被験者にレベチラセタム500mgを透析開始44時間前に単回経口投与したとき、レベチラセタムの非透析時の消失半減期は34.7時間であったが、透析中は2.3時間に短縮した。レベチラセタム及びucb L057の透析による除去効率は高く、81%及び87%であった18)。[7.2、9.2.2、13.2参照]
| 薬物動態パラメータ | レベチラセタム | ucb L057 |
| Cmax(μg/mL) | 18.7[8.1] | 8.84[7.0] |
| tmax(h) | 0.7(0.4-1.0) | 44.0(44.0-44.0) |
| t1/2(h) | 34.7(29.2-38.6) | − |
| AUC0-44h(μg・h/mL) | 462[10.5] | 230[7.8] |
| CL/F(mL/min/1.73m2) | 10.9(9.4-13.1) | − |
| ダイアライザーの除去効率(%) | 81[7.5] | 87[7.2] |
| 血液透析中の消失半減期(h) | 2.3(2.1-2.6) | 2.1(1.9-2.6) |
| 血液透析クリアランス(mL/min) | 115.4[8.1] | 122.9[7.1] |
16.6.3 肝機能障害患者
軽度及び中等度(Child-Pugh分類A及びB)の成人肝機能低下者にレベチラセタムを単回経口投与したとき、レベチラセタムの全身クリアランスに変化はみられなかった。重度(Child-Pugh分類C)の肝機能低下者では、全身クリアランスが健康成人の約50%となった19)20)(外国人データ)。[7.3、9.3.1参照]
| 薬物動態パラメータ | 健康成人 (N=5) | 肝機能低下者 | ||
| Child-Pugh 分類A (N=5) | Child-Pugh 分類B (N=6) | Child-Pugh 分類C (N=5) | ||
| CLCR(mL/min/1.73m2)注4) | 93.1±13.8 | 120.8±11.9 | 99.6±13.2 | 63.5±13.5 |
| レベチラセタム | ||||
| Cmax(μg/mL) | 23.1±1.2 | 23.6±4.9 | 24.7±3.3 | 24.1±3.8 |
| tmax(h) | 0.8±0.3 | 0.6±0.2 | 0.5±0.0 | 1.6±1.5 |
| AUC(μg・h/mL) | 234±49 | 224±25 | 262±58 | 595±220 |
| t1/2(h) | 7.6±1.0 | 7.6±0.7 | 8.7±1.5 | 18.4±7.2 |
| CL/F(mL/min/1.73m2) | 63.4±9.7 | 62.5±8.7 | 55.4±10.5 | 29.2±13.5 |
16.6.4 高齢者
16.7 薬物相互作用
16.7.1 フェニトイン
16.7.2 バルプロ酸ナトリウム
健康成人16例を対象に、バルプロ酸ナトリウムの定常状態下においてレベチラセタムを1500mg単回経口投与したとき、バルプロ酸ナトリウムはレベチラセタムの薬物動態に影響を及ぼさなかった。レベチラセタムもバルプロ酸ナトリウムの薬物動態に影響を及ぼさなかった24)(外国人データ)。
16.7.3 経口避妊薬(エチニルエストラジオール及びレボノルゲストレルの合剤)
16.7.4 ジゴキシン
健康成人11例を対象に、ジゴキシン(1回0.25mgを1日1回)及びレベチラセタム1回1000mg1日2回7日間反復経口投与したとき、レベチラセタムはジゴキシンの薬物動態パラメータに影響を及ぼさなかった。ジゴキシンもレベチラセタムの薬物動態に影響を及ぼさなかった27)(外国人データ)。
16.7.5 ワルファリン
プロトロンビン時間の国際標準比(INR)を目標値の範囲内に維持するよう、ワルファリンの投与を継続的に受けている健康成人26例を対象に、ワルファリン(2.5〜7.5mg/日)及びレベチラセタム1回1000mg1日2回7日間反復経口投与したとき、レベチラセタムはワルファリン濃度に影響を及ぼさず、プロトロンビン時間も影響を受けなかった。ワルファリンもレベチラセタムの薬物動態に影響を及ぼさなかった28)(外国人データ)。
16.7.6 プロベネシド
健康成人23例を対象に、プロベネシド(1回500mgを1日4回)及びレベチラセタム1回1000mg1日2回4日間反復経口投与したとき、プロベネシドはレベチラセタムの薬物動態には影響を及ぼさなかったが、主代謝物ucb L057の腎クリアランスを61%低下させた29)(外国人データ)。
16.8 その他
16.8.1 生物学的同等性
<イーケプラ錠500mg、イーケプラドライシロップ50%>
健康成人26例にレベチラセタム500mg(ドライシロップ50%を1g又は500mg錠を1錠)を空腹時単回投与したとき、レベチラセタムの薬物動態パラメータは以下のとおりであった。ドライシロップ50%と500mg錠は生物学的に同等であることが確認された30)。
| 薬物動態パラメータ | ドライシロップ (N=26) | 錠剤 (N=26) | 製剤間の比較 幾何平均比注5) (90%信頼区間) |
| Cmax(μg/mL) | 20.9[24.5] | 19.6[28.1] | 1.0680 (0.9689,1.1772) |
| AUC0-t(μg・h/mL) | 149[15.6] | 151[15.2] | 0.9871 (0.9701,1.0044) |
| tmax(h) | 0.500(0.233-1.50) | 0.633(0.250-2.00) | − |
17. 臨床成績
17.1 有効性及び安全性に関する試験
<てんかん患者の部分発作(二次性全般化発作を含む)>
17.1.1 国内第III相試験(成人、単剤療法)
最近てんかんと診断された部分発作を有する16歳以上の患者を対象として、レベチラセタム1000〜2000mg/日(1000mg/日を投与中に発作がみられた場合は2000mg/日に増量)又は3000mg/日(発作の有無にかかわらず、3000mg/日に強制漸増)を単剤にて経口投与したとき、主要評価項目である1000〜2000mg/日群の最終評価用量における6ヵ月間発作消失患者の割合は、73.8%(45/61例)であった。1000〜2000mg/日群の最終評価用量での1年間発作消失患者の割合は59.0%(36/61例)であった。また、3000mg/日群における6ヵ月間発作消失患者の割合は22.2%(2/9例)、1年間発作消失患者の割合は11.1%(1/9例)であった31)。
副作用発現頻度は54.9%(39/71例)であった。主な副作用は傾眠32.4%(23/71例)であった。
副作用発現頻度は54.9%(39/71例)であった。主な副作用は傾眠32.4%(23/71例)であった。
17.1.2 国内第II/III相試験(成人、併用療法)
レベチラセタム1000mg/日、3000mg/日及びプラセボを12週間経口投与(既存の抗てんかん薬との併用)した場合、主要評価項目である週あたりの部分発作回数減少率は下表のとおりであり、プラセボ群とレベチラセタム群(1000及び3000mg/日)並びにレベチラセタム1000mg/日群の間で統計学的な有意差が認められた(それぞれp<0.001並びにp=0.006、投与群を因子、観察期間における対数変換した週あたりの部分発作回数を共変量とする共分散分析)。なお、各群における50%レスポンダーレート(週あたりの部分発作回数が観察期間と比べて50%以上改善した患者の割合)は、プラセボ群13.8%(9/65例)、1000mg/日群31.3%(20/64例)、3000mg/日群28.6%(18/63例)であった32)。
| 例数注1) | 週あたりの部分発作回数注2) | プラセボ群に対する減少率注3),注4) [95%信頼区間] (p値) | ||||
| 観察期間 | 評価期間 | 減少率(%) | ||||
| プラセボ群 | 65 | 2.73 | 2.67 | 6.11 | / | / |
| 1000mg/日群 | 64 | 3.58 | 2.25 | 19.61 | 20.9 [10.2,30.4] (p<0.001) | 18.8 [6.0,29.9] (p=0.006) |
| 3000mg/日群 | 63 | 3.44 | 2.08 | 27.72 | 23.0 [10.7,33.6] | |
増量期間及び評価期間の副作用発現頻度は1000mg/日投与群で56.9%(41/72例)、3000mg/日投与群で54.9%(39/71例)であった。主な副作用は1000mg/日投与群で、傾眠13.9%(10/72例)、鼻咽頭炎8.3%(6/72例)、浮動性めまい8.3%(6/72例)、3000mg/日投与群で傾眠9.9%(7/71例)、鼻咽頭炎8.5%(6/71例)、浮動性めまい5.6%(4/71例)であった。
17.1.3 国内第III相試験(成人、併用療法)
レベチラセタム500mg/日、1000mg/日、2000mg/日、3000mg/日及びプラセボを12週間経口投与(既存の抗てんかん薬との併用)した場合、評価期間における観察期間からの週あたりの部分発作回数減少率(中央値)は、それぞれ12.92%、18.00%、11.11%、31.67%及び12.50%であり、主要評価項目であるレベチラセタム1000mg/日群、3000mg/日群及びプラセボ群の3群間での評価期間における観察期間からの週あたりの部分発作回数減少率に、統計学的な有意差は認められなかった(p=0.067、Kruskal-Wallis検定)。なお、各群における50%レスポンダーレートは、プラセボ群11.6%(8/69例)、500mg/日群19.1%(13/68例)、1000mg/日群17.6%(12/68例)、2000mg/日群16.2%(11/68例)、3000mg/日群33.3%(22/66例)であった33)。
増量期間及び評価期間の副作用発現頻度は、500mg/日投与群60.6%(43/71例)、1000mg/日投与群61.4%(43/70例)、2000mg/日投与群58.6%(41/70例)、3000mg/日投与群64.3%(45/70例)であった。主な副作用は500mg/日投与群で、鼻咽頭炎14.1%(10/71例)、下痢9.9%(7/71例)、浮動性めまい7.0%(5/71例)、傾眠7.0%(5/71例)、1000mg/日投与群で、鼻咽頭炎18.6%(13/70例)、傾眠10.0%(7/70例)、2000mg/日投与群で、傾眠17.1%(12/70例)、鼻咽頭炎15.7%(11/70例)、挫傷7.1%(5/70例)、3000mg/日投与群で鼻咽頭炎21.4%(15/70例)、傾眠17.1%(12/70例)、好中球数減少7.1%(5/70例)であった。
増量期間及び評価期間の副作用発現頻度は、500mg/日投与群60.6%(43/71例)、1000mg/日投与群61.4%(43/70例)、2000mg/日投与群58.6%(41/70例)、3000mg/日投与群64.3%(45/70例)であった。主な副作用は500mg/日投与群で、鼻咽頭炎14.1%(10/71例)、下痢9.9%(7/71例)、浮動性めまい7.0%(5/71例)、傾眠7.0%(5/71例)、1000mg/日投与群で、鼻咽頭炎18.6%(13/70例)、傾眠10.0%(7/70例)、2000mg/日投与群で、傾眠17.1%(12/70例)、鼻咽頭炎15.7%(11/70例)、挫傷7.1%(5/70例)、3000mg/日投与群で鼻咽頭炎21.4%(15/70例)、傾眠17.1%(12/70例)、好中球数減少7.1%(5/70例)であった。
17.1.4 国内長期継続投与試験
国内第II/III相試験(成人、併用療法)を完了した患者151例を対象として、レベチラセタム1000〜3000mg/日を1日2回に分けて経口投与したときの部分発作回数は以下のとおりであった34)35)36)。
本試験に参加した被験者のうち、76例がその後計画された継続試験に移行し本試験を終了した(24〜36ヵ月で1例、36〜48ヵ月で47例、48ヵ月以降で28例)。
本試験に参加した被験者のうち、76例がその後計画された継続試験に移行し本試験を終了した(24〜36ヵ月で1例、36〜48ヵ月で47例、48ヵ月以降で28例)。
副作用発現頻度は92.1%(139/151例)であった。主な副作用は鼻咽頭炎55.6%(84/151例)、頭痛24.5%(37/151例)、傾眠22.5%(34/151例)であった。
17.1.5 国内第III相試験(小児)
既存の抗てんかん薬で十分な発作抑制効果が得られない部分発作を有する4歳以上16歳未満の小児てんかん患者73例を対象として、レベチラセタム40又は60mg/kg/日(体重50kg以上は2000又は3000mg/日)を1日2回に分けて14週間経口投与(既存の抗てんかん薬との併用)したとき、主要評価項目である観察期間からの週あたりの部分発作回数減少率の中央値(95%信頼区間)は、43.21%(26.19%,52.14%)であり、発作頻度の減少が認められた。
また、小児てんかん患者55例に14週以降もレベチラセタム20〜60mg/kg/日(体重50kg以上は1000〜3000mg/日)を1日2回に分けて継続投与したときの部分発作回数は以下のとおりであった37)。
また、小児てんかん患者55例に14週以降もレベチラセタム20〜60mg/kg/日(体重50kg以上は1000〜3000mg/日)を1日2回に分けて継続投与したときの部分発作回数は以下のとおりであった37)。
副作用発現頻度は58.9%(43/73例)であった。主な副作用は、傾眠42.5%(31/73例)であった。
<他の抗てんかん薬で十分な効果が認められないてんかん患者の強直間代発作に対する抗てんかん薬との併用療法>
17.1.6 国際共同第III相試験(成人)
既存の抗てんかん薬で十分な発作抑制効果が得られない強直間代発作を有する16歳以上のてんかん患者251例(日本人43例を含む)を対象として、レベチラセタム1000若しくは3000mg/日(1000mg/日から投与を開始し、投与8週までに発作がみられた場合は2週間隔で1000mg/日ずつ3000mg/日に増量)又はプラセボを28週間経口投与(既存の抗てんかん薬との併用)したとき、主要評価項目である観察期間からの週あたりの強直間代発作回数減少率は下表のとおりであり、プラセボ群とレベチラセタム群の間で統計学的な有意差が認められた(p<0.0001、投与群及び国を因子、観察期間における週あたりの強直間代発作回数を共変量とする共分散分析)38)。
| 例数注5) | 週あたりの強直間代発作回数注6) | プラセボ群との差注7) [95%信頼区間] (p値) | |||
| 観察期間 | 治療期間 | 減少率(%) | |||
| プラセボ群 | 109 | 0.83 | 0.65 | 19.64 | 56.13 [44.02,68.24] (p<0.0001) |
| レベチラセタム群 | 117 | 0.89 | 0.16 | 76.98 | |
副作用発現頻度は23.8%(30/126例)であった。主な副作用は傾眠2.4%(3/126例)であった。また、主な臨床検査値異常(副作用)は、尿中蛋白陽性7.1%(9/126例)、血小板数減少4.0%(5/126例)、好中球数減少3.2%(4/126例)であった。
17.1.7 国内第III相試験(小児)
既存の抗てんかん薬で十分な発作抑制効果が得られない強直間代発作を有する4歳以上16歳未満の小児てんかん患者13例を対象として、レベチラセタム40又は60mg/kg/日(体重50kg以上は2000又は3000mg/日)を24週間経口投与(既存の抗てんかん薬との併用)したとき、主要評価項目である観察期間からの週あたりの強直間代発作回数減少率の中央値(95%信頼区間)は、56.52%(−15.74%,98.18%)であった39)。
副作用発現頻度は38.5%(5/13例)であった。副作用は傾眠23.1%(3/13例)、運動緩慢7.7%(1/13例)、頭痛7.7%(1/13例)、下痢7.7%(1/13例)であった。また、臨床検査値異常(副作用)は、心電図QT延長7.7%(1/13例)であった。
副作用発現頻度は38.5%(5/13例)であった。副作用は傾眠23.1%(3/13例)、運動緩慢7.7%(1/13例)、頭痛7.7%(1/13例)、下痢7.7%(1/13例)であった。また、臨床検査値異常(副作用)は、心電図QT延長7.7%(1/13例)であった。
17.1.8 長期継続投与試験(成人及び小児)
国際共同第III相試験若しくは小児国内第III相試験を完了、又は国際共同第III相試験を効果不十分のため投与20週以降に中止した日本人患者44例を対象として、成人(16歳以上)ではレベチラセタム1000〜3000mg/日、小児ではレベチラセタム20〜60mg/kg/日(体重50kg以上は1000〜3000mg/日)を経口投与したとき、強直間代発作回数は以下のとおりであった40)41)。
副作用発現頻度は38.6%(17/44例)であった。主な副作用は傾眠11.4%(5/44例)であった。また、臨床検査値異常(副作用)は、心電図QT延長4.5%(2/44例)、アラニンアミノトランスフェラーゼ増加2.3%(1/44例)、アスパラギン酸アミノトランスフェラーゼ増加2.3%(1/44例)、C-反応性蛋白増加2.3%(1/44例)、体重増加2.3%(1/44例)であった。
18. 薬効薬理
18.1 作用機序
18.2 てんかん発作に対する作用
18.3 抗てんかん原性作用
扁桃核電気刺激キンドリングラットにおいて、キンドリング形成を抑制した52)。
18.4 中枢神経に対するその他の作用
19. 有効成分に関する理化学的知見
19.1. レベチラセタム
| 一般的名称 | レベチラセタム |
|---|---|
| 一般的名称(欧名) | Levetiracetam |
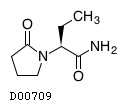
| 化学名 | (2S)-2-(2-Oxopyrrolidine-1-yl)butyramide |
| 分子式 | C8H14N2O2 |
| 分子量 | 170.21 |
| 物理化学的性状 | 白色〜淡灰白色の結晶性の粉末である。水に極めて溶けやすく、メタノール又はエタノール(99.5)に溶けやすく、アセトニトリル、アセトン又は2-プロパノールにやや溶けやすく、トルエン又はジエチルエーテルに溶けにくく、ヘキサンにほとんど溶けない。 |
| KEGG DRUG |
|
22. 包装
バラ[乾燥剤入り]
100g
23. 主要文献
- 日本てんかん学会:抗てんかん発作薬を服用しているてんかんのある人において、自動車運転や危険を伴う機械操作を行う際の留意事項(2026年3月17日)
- 厚生労働省:重篤副作用疾患別対応マニュアル 薬剤性過敏症症候群
- 日本人健康成人におけるレベチラセタム単回投与時の薬物動態(イーケプラ錠:2010年7月23日承認、申請資料概要2.7.6.3.1)
- 日本人健康成人におけるレベチラセタム反復投与時の薬物動態(イーケプラ錠:2010年7月23日承認、申請資料概要2.7.6.3.2)
- 日本人健康成人におけるレベチラセタム錠及び注射剤の単回投与時の比較(イーケプラ点滴静注:2014年7月4日承認、申請資料概要2.7.6.1.1)
- 外国小児てんかん患者におけるレベチラセタム単回投与時の薬物動態[1](イーケプラ錠/ドライシロップ:2013年5月31日承認、申請資料概要2.7.6.2.1)
- 外国小児てんかん患者におけるレベチラセタム単回投与時の薬物動態[2](イーケプラ錠/ドライシロップ:2013年5月31日承認、審査報告書)
- レベチラセタムに関する母集団薬物動態解析-1(イーケプラ錠:2010年7月23日承認、申請資料概要2.7.2.2.3)
- Toublanc,N.et al., Drug Metab.Pharmacokinet., 29, 61-68, (2014) »PubMed
- 大國壽他, 診療と新薬, 58 (9), 667-674, (2021)
- 日本人健康成人におけるレベチラセタムの薬物動態に及ぼす食事の影響(イーケプラ錠:2010年7月23日承認、申請資料概要2.7.6.1.2)
- Ramael,S.et al., Clin.Ther., 28, 734-744, (2006) »PubMed
- Strolin Benedetti,M.et al., Eur.J.Clin.Pharmacol., 59, 621-630, (2003) »PubMed
- 分布(イーケプラ錠:2010年7月23日承認、申請資料概要2.6.4.1,2.6.4.4)
- 代謝(代謝経路)(イーケプラ錠:2010年7月23日承認、申請資料概要2.4.3.3,2.6.2.2)
- 代謝(薬物動態学的薬物相互作用)(イーケプラ錠:2010年7月23日承認、申請資料概要2.6.4.7)
- 排泄(イーケプラ錠:2010年7月23日承認、申請資料概要2.7.2.3.2)
- Yamamoto,J.et al., Clin.Drug Investig., 34, 819-828, (2014) »PubMed
- Brockmoller,J.et al., Clin.Pharmacol.Ther., 77, 529-541, (2005) »PubMed
- 肝機能障害患者(イーケプラ錠:2010年7月23日承認、申請資料概要2.7.6.4.5)
- 高齢者(外国人)におけるレベチラセタム単回及び反復経口投与時の薬物動態(イーケプラ錠:2010年7月23日承認、申請資料概要2.5.3.4,2.7.6.4.1)
- Browne,T.R.et al., J.Clin.Pharmacol., 40, 590-595, (2000) »PubMed
- 薬物相互作用(フェニトイン)(イーケプラ錠:2010年7月23日承認、申請資料概要2.7.6.5.6)
- Coupez,R.et al., Epilepsia, 44, 171-178, (2003) »PubMed
- Ragueneau-Majlessi,I.et al., Epilepsia, 43, 697-702, (2002) »PubMed
- 薬物相互作用(経口避妊薬)(イーケプラ錠:2010年7月23日承認、申請資料概要2.7.6.5.1)
- Levy,R.H.et al., Epilepsy Res., 46, 93-99, (2001) »PubMed
- Ragueneau-Majlessi,I.et al., Epilepsy Res., 47, 55-63, (2001) »PubMed
- レベチラセタム及び代謝物の薬物動態に及ぼすプロベネシドの影響(イーケプラ錠:2010年7月23日承認、申請資料概要2.7.6.5.4)
- ドライシロップと錠剤の生物学的同等性試験(イーケプラ錠/ドライシロップ:2013年5月31日承認、申請資料概要2.7.6.1.1)
- 日本における部分発作単剤療法の第III相試験(イーケプラ錠/ドライシロップ/点滴静注:2015年2月20日承認、審査報告書)
- 日本における部分発作併用療法のプラセボ対照比較試験(国内第II/III相試験(成人、併用療法))(イーケプラ錠:2010年7月23日承認、申請資料概要2.7.6.7.1)
- 日本における部分発作併用療法のプラセボ対照比較試験(国内第III相試験(成人、併用療法))(イーケプラ錠:2010年7月23日承認、申請資料概要2.7.6.7.2)
- 八木和一他, てんかん研究, 29, 441-454, (2012) »DOI
- 国内長期継続投与試験[1](イーケプラ錠:2010年7月23日承認、申請資料概要2.7.6.8.1)
- 国内長期継続投与試験[2](イーケプラ錠:2010年7月23日承認、審査報告書)
- 日本における部分発作併用療法の小児第III相試験(イーケプラ錠/ドライシロップ:2013年5月31日承認、申請資料概要2.7.6.4.2)
- 日本及び中国における強直間代発作併用療法のプラセボ対照比較試験(イーケプラ錠/ドライシロップ/点滴静注:2016年2月29日承認、申請資料概要2.7.6.1.1)
- 日本における強直間代発作併用療法の小児第III相試験(イーケプラ錠/ドライシロップ/点滴静注:2016年2月29日承認、申請資料概要2.7.6.2.1)
- 日本における強直間代発作併用療法の長期継続投与試験[1](イーケプラ錠/ドライシロップ/点滴静注:2016年2月29日承認、申請資料概要2.7.6.2.2)
- 日本における強直間代発作併用療法の長期継続投与試験[2](イーケプラ錠/ドライシロップ/点滴静注:2016年2月29日承認、審査報告書)
- Noyer,M.et al., Eur.J.Pharmacol., 286, 137-146, (1995) »PubMed
- Lynch,B.A.et al., Proc.Nat.Acad.Sci.U.S.A., 101, 9861-9866, (2004)
- Lukyanetz,E.A.et al., Epilepsia,, 43, 9-18, (2002) »PubMed
- Pisani,A.et al., Epilepsia, 45, 719-728, (2004) »PubMed
- Rigo,J.M.et al., Br.J.Pharmacol., 136, 659-672, (2002) »PubMed
- Margineanu,D.G.et al., Pharmacol.Res., 42, 281-285, (2000) »PubMed
- Kaminski,R.M.et al., Neuropharmacology, 54, 715-720, (2008) »PubMed
- Klitgaard,H.et al., Eur.J.Pharmacol., 353, 191-206, (1998) »PubMed
- Gower,A.J.et al., Epilepsy Res., 22, 207-213, (1995) »PubMed
- Gower,A.J.et al., Eur.J.Pharmacol., 222, 193-203, (1992) »PubMed
- Loscher,W.et al., J.Pharmacol.Exp.Ther., 284, 474-479, (1998) »PubMed
- Lamberty,Y.et al., Epilepsy Behav., 1, 333-342, (2000) »PubMed
- Hanon,E.et al., Seizure, 10, 287-293, (2001) »PubMed
24. 文献請求先及び問い合わせ先
文献請求先
沢井製薬株式会社
医薬品情報センター
〒532-0003
大阪市淀川区宮原5丁目2-30
電話:0120-381-999
FAX:06-7708-8966
製品情報問い合わせ先
沢井製薬株式会社
医薬品情報センター
〒532-0003
大阪市淀川区宮原5丁目2-30
電話:0120-381-999
FAX:06-7708-8966
26. 製造販売業者等
26.1 製造販売元
沢井製薬株式会社
大阪市淀川区宮原5丁目2-30