医薬品情報
| 総称名 | レットヴィモ |
|---|---|
| 一般名 | セルペルカチニブ |
| 欧文一般名 | Selpercatinib |
| 製剤名 | セルペルカチニブカプセル |
| 薬効分類名 | 抗悪性腫瘍剤 RET注2)受容体型チロシンキナーゼ阻害剤 注2)RET:rearranged during transfection |
| 薬効分類番号 | 4291 |
| ATCコード | L01EX22 |
| KEGG DRUG |
D11713
セルペルカチニブ
|
| JAPIC | 添付文書(PDF) |
添付文書情報2026年2月 改訂(第9版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| レットヴィモカプセル40mg | Retevmo Capsules | 日本イーライリリー | 4291075M1027 | 4066.2円/カプセル | 劇薬, 処方箋医薬品注1) |
| レットヴィモカプセル80mg | Retevmo Capsules | 日本イーライリリー | 4291075M2023 | 7717.4円/カプセル | 劇薬, 処方箋医薬品注1) |
1. 警告
本剤は、緊急時に十分対応できる医療施設において、がん化学療法に十分な知識・経験を持つ医師のもとで、本剤の投与が適切と判断される症例についてのみ投与すること。また、治療開始に先立ち、患者又はその家族に有効性及び危険性を十分説明し、同意を得てから投与すること。
2. 禁忌
次の患者には投与しないこと
本剤の成分に対しアナフィラキシー等の重篤な過敏症の既往歴のある患者
4. 効能または効果
○RET融合遺伝子陽性の進行・再発の固形腫瘍
○RET遺伝子変異陽性の根治切除不能な甲状腺髄様癌
5. 効能または効果に関連する注意
<RET融合遺伝子陽性の進行・再発の非小細胞肺癌>
5.1 十分な経験を有する病理医又は検査施設における検査により、RET融合遺伝子陽性が確認された患者に投与すること。検査にあたっては、承認された体外診断用医薬品又は医療機器注)を用いること。
5.2 「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、本剤以外の治療の実施についても慎重に検討し、適応患者の選択を行うこと。[17.1.1参照]
5.3 本剤の術後補助療法における有効性及び安全性は確立していない。
<RET融合遺伝子陽性の進行・再発の甲状腺癌>
5.4 十分な経験を有する病理医又は検査施設における検査により、RET融合遺伝子陽性が確認された患者に投与すること。検査にあたっては、承認された体外診断用医薬品又は医療機器注)を用いること。
5.5 放射性ヨウ素による治療の適応となる患者においては、当該治療を優先すること。
<RET融合遺伝子陽性の進行・再発の固形腫瘍(非小細胞肺癌及び甲状腺癌を除く)>
5.6 十分な経験を有する病理医又は検査施設における検査により、RET融合遺伝子陽性が確認された患者に投与すること。検査にあたっては、承認された体外診断用医薬品又は医療機器注)を用いること。
5.7 組織球症患者は本剤の投与対象となり得る。
5.8 臨床試験に組み入れられた患者のがん種等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、本剤以外の治療の実施についても慎重に検討し、適応患者の選択を行うこと。[17.1.1参照]
5.9 本剤の手術の補助療法における有効性及び安全性は確立していない。
<RET遺伝子変異陽性の根治切除不能な甲状腺髄様癌>
5.10 十分な経験を有する病理医又は検査施設における検査により、RET遺伝子変異が確認された患者に投与すること。生殖細胞系列のRET遺伝子変異が陰性又は不明の場合は、承認された体外診断用医薬品又は医療機器注)を用いて検査を行うこと。
5.11 「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、本剤以外の治療の実施についても慎重に検討し、適応患者の選択を行うこと。[17.1.1参照]
注)承認された体外診断用医薬品又は医療機器に関する情報については、以下のウェブサイトから入手可能である:
https://www.pmda.go.jp/review-services/drug-reviews/review-information/cd/0001.html
6. 用法及び用量
通常、成人にはセルペルカチニブとして1回160mgを1日2回経口投与する。なお、患者の状態により適宜減量する。
通常、12歳以上の小児には体表面積に合わせて次の投与量(セルペルカチニブとして1回約92mg/m2)を1日2回経口投与する。なお、患者の状態により適宜減量する。
| 体表面積 | 1回投与量 |
| 1.2m2未満 | 80mg |
| 1.2m2以上1.6m2未満 | 120mg |
| 1.6m2以上 | 160mg |
7. 用法及び用量に関連する注意
7.1 他の抗悪性腫瘍剤との併用について、有効性及び安全性は確立していない。
7.2 本剤投与により副作用が発現した場合には、以下の基準を考慮して、休薬・減量・中止すること。
| 減量レベル | 投与量 |
| 通常投与量 | 1回160mg 1日2回 |
| 1段階減量 | 1回120mg 1日2回 |
| 2段階減量 | 1回80mg 1日2回 |
| 3段階減量 | 1回40mg 1日2回 |
| 体表面積 | 減量レベル | 投与量 |
| 1.2m2未満 | 通常投与量 | 1回80mg 1日2回 |
| 1段階減量 | 1回40mg/1回80mg 1日2回 (1日量120mg) | |
| 2段階減量 | 1回40mg 1日2回 | |
| 3段階減量注1) | 1回40mg 1日1回 | |
| 1.2m2以上1.6m2未満 | 通常投与量 | 1回120mg 1日2回 |
| 1段階減量 | 1回80mg 1日2回 | |
| 2段階減量 | 1回40mg/1回80mg 1日2回 (1日量120mg) | |
| 3段階減量注1) | 1回40mg 1日1回 | |
| 1.6m2以上 | 通常投与量 | 1回160mg 1日2回 |
| 1段階減量 | 1回120mg 1日2回 | |
| 2段階減量 | 1回80mg 1日2回 | |
| 3段階減量 | 1回40mg 1日2回 |
| 副作用 | 程度注2) | 処置 |
| ALT又はAST増加 | グレード3又は4 | グレード1以下に回復するまで休薬し、回復後は2段階減量して投与再開できる。 再開後に2週間以上再発しない場合には、1段階増量することができる。更に4週間以上再発しない場合には、もう1段階増量することができる。 減量した用量で投与中に再発した場合には、中止する。 |
| QT間隔延長 | QTc間隔>500msec | QTc間隔<470msecに回復するまで休薬し、回復後は1段階減量して投与再開できる。 2段階減量した用量で投与中に再発した場合には、中止する。 |
| 重篤な不整脈を疑う所見や症状が認められた場合 | 中止する。 | |
| 高血圧 | グレード3又は4 | 回復するまで休薬し、回復後は1段階減量して投与再開できる。 |
| 過敏症(アナフィラキシー等の重篤な過敏症を除く) [11.1.3参照] | グレード1〜4 | 回復するまで休薬し、副腎皮質ステロイドの全身投与を考慮する。回復後は副腎皮質ステロイドを併用しながら3段階減量して投与再開できる。 再開後に7日以上再発しない場合には、1段階ずつ発現時の用量まで増量できる。増量後に7日以上再発しない場合には、副腎皮質ステロイドを漸減する。 |
| 間質性肺疾患 | グレード2 | 回復するまで休薬し、回復後は1段階減量して投与再開できる。 |
| グレード3又は4 | 中止する。 | |
| 上記以外の副作用 | グレード3又は4 | 回復するまで休薬し、回復後は1段階減量して投与再開できる。 |
8. 重要な基本的注意
8.1 肝機能障害があらわれることがあるので、本剤の投与開始前及び投与期間中は定期的に肝機能検査を行い、患者の状態を十分に観察すること。[11.1.1参照]
8.2 QT間隔延長があらわれることがあるので、本剤の投与開始前には患者のQTc間隔が470msec以下であることを確認するとともに血清電解質検査(カリウム、マグネシウム等)を行うこと。心電図及び血清電解質検査を投与開始後1週間時点及び投与開始後6ヵ月間は毎月1回行い、以降も必要に応じて行うこと。また、必要に応じて電解質補正を行うこと。[11.1.2、17.3.1参照]
8.3 高血圧があらわれることがあるので、本剤の投与開始前に血圧が適切に管理されていることを確認すること。本剤投与中は定期的に血圧を測定すること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.3 肝機能障害患者
9.3.1 重度の肝機能障害のある患者(Child-Pugh分類C)
減量を考慮するとともに、患者の状態をより慎重に観察し、副作用の発現に十分注意すること。本剤の血中濃度が上昇し、副作用が増強されるおそれがある。[16.6.2参照]
9.4 生殖能を有する者
9.4.1 妊娠する可能性のある女性には、本剤投与中及び最終投与後1ヵ月間において避妊する必要性及び適切な避妊法について説明すること。[9.5参照]
9.4.2 男性には、本剤投与中及び最終投与後1週間においてバリア法(コンドーム)を用いて避妊する必要性について説明すること。[15.2.1参照]
9.4.3 成長期にある若年男性又は男児に投与する場合には、造精機能の低下があらわれる可能性があることを考慮すること。幼若ラットにおいて、精巣の精上皮変性、精巣上体の精子枯渇、精子運動率低値、異常形態精子比率高値及び受胎能の低下が認められ、精巣及び精巣上体の所見に回復性は認められていない1)。
9.5 妊婦
9.6 授乳婦
授乳しないことが望ましい。乳汁移行に関するデータはないが、本剤はBCRPの基質であるため、乳汁移行の可能性がある。
9.7 小児等
9.7.1 成長期にある若年者においては、骨成長について以下の点に注意すること。
・骨端線に異常がないか十分に観察すること。骨端線に異常が認められた場合には、投与継続の可否を慎重に判断すること。
・関節痛及び歩行障害について十分に観察すること。大腿骨頭すべり症等の骨端離開があらわれることがある。
[15.2.2参照]
9.7.2 12歳未満の小児等を対象とした臨床試験は実施していない。
10. 相互作用
相互作用序文
本剤は、主にCYP3A4によって代謝され、CYP2C8及び3Aの阻害作用を示す。また、本剤の溶解度はpHの上昇により低下する。
薬物代謝酵素用語
CYP3A4
薬物代謝酵素用語
CYP2C8
薬物代謝酵素用語
CYP3A
10.2 併用注意
| CYP2C8の基質となる薬剤 レパグリニド ピオグリタゾン モンテルカスト等 [16.7.6参照] | これらの薬剤の副作用が増強されるおそれがあるので、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | 本剤がCYP2C8を阻害することにより、これらの薬剤の血中濃度が上昇する可能性がある。 |
| CYP3Aの基質となる薬剤 ミダゾラム トリアゾラム ロミタピド等 [16.7.5参照] | これらの薬剤の副作用が増強されるおそれがあるので、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | 本剤がCYP3Aを阻害することにより、これらの薬剤の血中濃度が上昇する可能性がある。 |
| CYP3A阻害剤 イトラコナゾール クラリスロマイシン エリスロマイシン等 [16.7.1、16.7.2参照] | 本剤の副作用が増強されるおそれがあるので、これらの薬剤との併用は可能な限り避けること。やむを得ず併用する場合には、本剤の減量を考慮するとともに、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | これらの薬剤がCYP3Aを阻害することにより、本剤の血中濃度が上昇する可能性がある。 |
| CYP3A誘導剤 リファンピシン フェニトイン ボセンタン等 [16.7.3、16.7.4参照] | 本剤の有効性が減弱するおそれがあるので、これらの薬剤との併用は可能な限り避け、CYP3A誘導作用のない薬剤への代替を考慮すること。 | これらの薬剤等がCYP3Aを誘導することにより、本剤の血中濃度が低下する可能性がある。 |
| セイヨウオトギリソウ(St.John's Wort、セント・ジョーンズ・ワート)含有製品 | 本剤の有効性が減弱するおそれがあるので、摂取しないよう注意すること。 | これらの薬剤等がCYP3Aを誘導することにより、本剤の血中濃度が低下する可能性がある。 |
| プロトンポンプ阻害剤 オメプラゾール ランソプラゾール エソメプラゾール等 [16.7.7参照] | 本剤の有効性が減弱するおそれがあるので、これらの薬剤との併用は可能な限り避けること。やむを得ず併用する場合には、本剤とともに食後に投与すること。 | これらの薬剤による胃内pHの上昇により、本剤の吸収が低下し、本剤の血中濃度が低下する可能性がある。 |
| H2受容体拮抗剤 ラニチジン ファモチジン シメチジン等 [16.7.8参照] | 本剤の有効性が減弱するおそれがあるので、これらの薬剤との併用は可能な限り避けること。やむを得ず併用する場合には、本剤と服用時間をずらすこと(ラニチジンを本剤投与10時間前及び2時間後に投与したときの本剤の血中濃度への影響は限定的であった)。 | これらの薬剤による胃内pHの上昇により、本剤の吸収が低下し、本剤の血中濃度が低下する可能性がある。 |
| 制酸剤 炭酸カルシウム 水酸化マグネシウム 水酸化アルミニウム等 | 本剤の有効性が減弱するおそれがあるので、これらの薬剤との併用は可能な限り避けること。やむを得ず併用する場合には、本剤と服用時間をずらすこと(制酸剤を本剤投与2時間前又は2時間後に投与したときの本剤の血中濃度への影響は限定的であった)。 | これらの薬剤による胃内pHの上昇により、本剤の吸収が低下し、本剤の血中濃度が低下する可能性がある。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 肝機能障害(36.4%)[8.1参照]
11.1.3 過敏症(5.1%)
発疹、発熱等の症状を伴う遅発性の過敏症があらわれることがある。[7.2参照]
11.1.4 高血圧(30.0%)[9.1.2参照]
11.2 その他の副作用
| 20%以上 | 5〜20%未満 | 5%未満 | 頻度不明 | |
| 消化器 | 口内乾燥(34.2%)、下痢 | 便秘、悪心、口内炎、腹痛 | 嘔吐 | |
| 一般・全身及び投与部位反応 | 疲労 | 浮腫、発熱 | ||
| 呼吸器 | 鼻出血、肺炎 | |||
| 感染症 | 尿路感染 | |||
| 内分泌 | 甲状腺機能低下症 | |||
| 代謝・栄養障害 | 食欲減退 | |||
| 精神神経系 | 頭痛 | 浮動性めまい | ||
| 皮膚 | 発疹 | |||
| 生殖器 | 勃起不全 | |||
| 血液 | 血小板減少、白血球減少、好中球減少 | リンパ球減少、貧血 | ||
| 臨床検査値異常 | 血中クレアチニン増加 | 低マグネシウム血症、低カリウム血症、低カルシウム血症、低アルブミン血症 | ||
| その他 | 乳び胸、乳び腹水 |
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
15. その他の注意
15.2 非臨床試験に基づく情報
15.2.2 動物試験(成長板が閉鎖していない幼若ラット、若齢ラット及び若齢ミニブタ)において、本剤の反復投与により骨端成長板の異常(骨端軟骨の肥大、過形成及び異形成)が、ヒトに160mg1日2回の用量で投与したときの臨床曝露量よりも低い曝露量で認められている。また、幼若ラットにおいて、骨端成長板の変化に関連して、骨密度及び大腿骨長の低値が、ヒトに160mg1日2回の用量で投与したときの臨床曝露量のそれぞれ0.8倍及び1.9倍で認められている1)。[9.7.1参照]
15.2.3 ラットを用いた2年間がん原性試験において、ヒトに160mg1日2回の用量で投与したときの臨床曝露量に相当する用量で雌に腟腫瘍が認められている2)。
16. 薬物動態
16.1 血中濃度
16.1.1 単回及び反復投与
進行固形腫瘍患者94例に本剤160mgを単回経口投与したときの血漿中濃度推移及び薬物動態パラメータは以下のとおりであった3)(外国人データ)。
図1)本剤160mgを単回経口投与後(第1サイクル第1日目)の血漿中濃度推移(平均値±標準偏差)
| 例数 | 94 |
| Cmax(ng/mL) | 1120(85.7) |
| tmax注1)(hr) | 1.96(0.50-7.83) |
| AUCτ(ng・hr/mL) | 7430注2)(67.5) |
日本人の進行固形腫瘍患者58例に本剤160mgを1日2回反復経口投与したときの定常状態における血漿中濃度推移及び薬物動態パラメータは以下のとおりであった3)。
図2)本剤160mgを1日2回反復経口投与後(第1サイクル第8日目)の定常状態における血漿中濃度推移(平均値±標準偏差)
| 例数 | 58 |
| Cmax,ss(ng/mL) | 4060(34.4) |
| tmax,ss注3)(hr) | 2.08(0.00-8.10) |
| AUCτ,ss(ng・hr/mL) | 37100(39.3) |
| CLss/F(L/hr) | 4.31(39.3) |
血漿中濃度は反復投与後8日までに定常状態に到達した。また、本剤160mgを1日2回反復経口投与した際の投与8日目におけるセルペルカチニブの蓄積率は3.40であった。
16.2 吸収
16.2.1 絶対的バイオアベイラビリティ
健康成人6例に本剤160mgを単回経口投与したときの絶対的バイオアベイラビリティの幾何平均値は73.2%であった4)(外国人データ)。
16.2.2 食事の影響
健康成人20例に本剤160mgを高脂肪食摂取後に単回経口投与したとき、空腹時投与に対する食後投与におけるセルペルカチニブのCmax及びAUCinfの幾何平均値の比はそれぞれ0.862及び1.09であった5)(外国人データ)。
16.3 分布
16.3.1 蛋白結合率
セルペルカチニブのヒト血漿タンパク結合率は約96%であり、濃度依存性は認められなかった(in vitro)6)。
16.3.2 血液/血漿中濃度比
セルペルカチニブの血液/血漿中濃度比は約0.7であった(in vitro)7)。
16.4 代謝
16.5 排泄
健康成人6例に[14C]-セルペルカチニブ160mgを単回経口投与したとき、投与432時間後までに投与した放射能の約69%(未変化体は約14%)が糞便中に排泄され、約24%(未変化体は約11.5%)が尿中に排泄された4)(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
本剤160mgを単回経口投与したとき、腎機能正常被験者(10例)に対する軽度の腎機能障害患者(8例)の非結合形セルペルカチニブのCmax及びAUCinfの幾何平均値の比は、それぞれ1.30及び1.07であった。腎機能正常被験者(10例)に対する中等度の腎機能障害患者(8例)の非結合形セルペルカチニブのCmax及びAUCinfの幾何平均値の比は、それぞれ1.67及び1.89であった。腎機能正常被験者(10例)に対する重度の腎機能障害患者(7例)の非結合形セルペルカチニブのCmax及びAUCinfの幾何平均値の比は、それぞれ1.04及び1.54であった9)(外国人データ)。血液透析の有無によらず、末期腎不全患者に関するデータはない。
16.6.2 肝機能障害患者
本剤160mgを単回経口投与したとき、肝機能正常被験者(12例)に対する軽度の肝機能障害患者(8例)の非結合形セルペルカチニブのCmax及びAUCinfの幾何平均値の比は、それぞれ1.78及び1.33であった。肝機能正常被験者(12例)に対する中等度の肝機能障害患者(8例)の非結合形セルペルカチニブのCmax及びAUCinfの幾何平均値の比は、それぞれ0.989及び0.991であった。肝機能正常被験者(12例)に対する重度の肝機能障害患者(8例)の非結合形セルペルカチニブのCmax及びAUCinfの幾何平均値の比は、それぞれ2.32及び3.28であった10)(外国人データ)。[9.3.1参照]
16.6.3 小児
国際共同第I/II相試験(LIBRETTO-001試験及びLIBRETTO-121試験)に組み入れられた830例(小児患者24例を含む)のデータを用いて母集団薬物動態モデルが構築された。構築された母集団薬物動態モデルを用いて、仮想患者の体表面積及び体重情報に基づく薬物動態シミュレーションを実施した結果、[1]体表面積1.2m2未満の患者に80mg、[2]体表面積1.2m2以上1.6m2未満の患者に120mg、[3]体表面積1.6m2以上の患者に160mgをそれぞれ1日2回反復経口投与した際の、定常状態における本剤のCmax(ng/mL)及びAUC24h(ng・hr/mL)の中央値は、[1]2860及び47700、[2]3100及び54900並びに[3]3670及び66900と推定された11)。
16.7 薬物相互作用
16.7.1 イトラコナゾール
16.7.2 フルコナゾール、ジルチアゼム
16.7.3 リファンピシン
16.7.4 ボセンタン、モダフィニル
16.7.5 ミダゾラム
16.7.6 レパグリニド
16.7.7 オメプラゾール
健康成人20例にオメプラゾール(プロトンポンプ阻害剤)40mgを1日1回反復経口投与し、本剤160mgを空腹時に単回経口投与したとき、本剤単独投与時に対するオメプラゾール併用投与時のセルペルカチニブのCmax及びAUCinfの幾何平均値の比は、0.123及び0.313であった。また、オメプラゾールを反復経口投与し、本剤160mgを高脂肪食摂取後に単回経口投与したとき、本剤単独投与時に対するオメプラゾール併用投与時のセルペルカチニブのCmax及びAUCinfの幾何平均値の比は、0.586及び0.938であった。オメプラゾールを反復経口投与し、本剤160mgを低脂肪食摂取後に単回経口投与したとき、本剤単独投与時に対するオメプラゾール併用投与時のセルペルカチニブのCmax及びAUCinfの幾何平均値の比は、0.782及び1.00であった5)16)(外国人データ)。[10.2参照]
16.7.8 ラニチジン
16.7.9 その他
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国際共同第I/II相試験(LIBRETTO-001試験)
<RET融合遺伝子陽性の進行・再発の非小細胞肺癌及び甲状腺癌、RET遺伝子変異陽性の根治切除不能な甲状腺髄様癌>
[1]化学療法歴のあるRET融合遺伝子陽性の切除不能な進行・再発の非小細胞肺癌患者134例(日本人患者25例を含む)及び[2]化学療法歴のないRET融合遺伝子陽性の切除不能な進行・再発の非小細胞肺癌患者35例(日本人患者1例を含む)に本剤1回160mgを1日2回経口投与した。主要評価項目であるRECIST ver.1.1に基づく独立評価委員会判定による奏効率は、それぞれ[1]55.2%(95%信頼区間:46.4-63.8)及び[2]71.4%(95%信頼区間:53.7-85.4)であった。
12歳以上の[3]化学療法歴のあるRET融合遺伝子陽性の根治切除不能な甲状腺癌患者10例注1)(日本人患者1例を含む)、[4]化学療法歴のないRET融合遺伝子陽性の根治切除不能な甲状腺癌患者12例注2)、[5]化学療法歴のあるRET遺伝子変異陽性の根治切除不能な甲状腺髄様癌患者97例(日本人患者1例を含む)及び[6]化学療法歴のないRET遺伝子変異陽性の根治切除不能な甲状腺髄様癌患者90例に本剤1回160mg注3)を1日2回経口投与した。主要評価項目であるRECIST ver.1.1に基づく独立評価委員会判定による奏効率は、それぞれ[3]50.0%(95%信頼区間:18.7-81.3)、[4]100%(95%信頼区間:73.5-100)、[5]68.0%(95%信頼区間:57.8-77.1)及び[6]63.3%(95%信頼区間:52.5-73.2)であった3)。(2020年3月30日データカットオフ)[5.2、5.11参照]
12歳以上の[3]化学療法歴のあるRET融合遺伝子陽性の根治切除不能な甲状腺癌患者10例注1)(日本人患者1例を含む)、[4]化学療法歴のないRET融合遺伝子陽性の根治切除不能な甲状腺癌患者12例注2)、[5]化学療法歴のあるRET遺伝子変異陽性の根治切除不能な甲状腺髄様癌患者97例(日本人患者1例を含む)及び[6]化学療法歴のないRET遺伝子変異陽性の根治切除不能な甲状腺髄様癌患者90例に本剤1回160mg注3)を1日2回経口投与した。主要評価項目であるRECIST ver.1.1に基づく独立評価委員会判定による奏効率は、それぞれ[3]50.0%(95%信頼区間:18.7-81.3)、[4]100%(95%信頼区間:73.5-100)、[5]68.0%(95%信頼区間:57.8-77.1)及び[6]63.3%(95%信頼区間:52.5-73.2)であった3)。(2020年3月30日データカットオフ)[5.2、5.11参照]
注1)未分化癌2例、低分化癌2例を含む。
注2)低分化癌1例を含む。
注3)12歳以上の小児に対する本剤の承認用法・用量は、(1)体表面積1.2m2未満の患者に80mg、(2)体表面積1.2m2以上1.6m2未満の患者に120mg、(3)体表面積1.6m2以上の患者に160mgをそれぞれ1日2回経口投与である。
安全性評価対象400例に認められた主な副作用は、口内乾燥(35.3%)、高血圧(31.8%)、ALT増加(28.0%)、AST増加(26.8%)、疲労(25.3%)等であった。(2020年3月30日データカットオフ)
<RET融合遺伝子陽性の進行・再発の固形腫瘍(非小細胞肺癌及び甲状腺癌を除く)>
RET融合遺伝子陽性の進行・再発の固形腫瘍(非小細胞肺癌及び甲状腺癌を除く)患者として、本剤の忍容性の評価を目的とした[7]第I相パート(本剤20mgを1日1回又は本剤1回20、40、60、80、120、160、200若しくは240mgを1日2回経口投与)注4)の5例、本剤の有効性及び安全性を検討することを目的とした第II相パート(本剤1回160mgを1日2回経口投与)のうち、[8]化学療法歴のある患者を対象としたコホートの33例(日本人患者8例を含む)、[9]化学療法歴のない患者を対象としたコホートの2例、[10]腫瘍組織検体以外でRET融合遺伝子陽性が確認された患者等を対象としたコホートの12例(日本人患者3例を含む)が有効性の評価対象とされた。第II相パートの主要評価項目であるRECIST ver.1.1に基づく独立評価委員会判定による奏効率は、それぞれ[8]57.6%(95%信頼区間:39.2-74.5)、[9]0%及び[10]16.7%であった3)。(2023年1月13日データカットオフ)[5.8参照]
注4)承認された用法・用量は本剤1回160mgを1日2回経口投与である。
| がん種 | 奏効例数/評価例数 | 奏効率 |
| 大腸癌 | 4/13 | 30.8% |
| 膵癌注5) | 7/13 | 53.8% |
| 唾液腺癌注6) | 2/4 | 50.0% |
| 肉腫注7) | 1/3 | 33.3% |
| 胆道癌注6) | 1/3 | 33.3% |
| 原発不明癌 | 1/3 | 33.3% |
| 黄色肉芽腫注8) | 0/2 | 0% |
| 乳癌注6) | 2/2 | 100% |
| 皮膚癌注9) | 1/2 | 50.0% |
| カルチノイド | 1/1 | 100% |
| 小腸癌 | 1/1 | 100% |
| 直腸神経内分泌腫瘍注10) | 0/1 | 0% |
| 卵巣癌 | 1/1 | 100% |
| 肺癌肉腫 | 0/1 | 0% |
| 神経内分泌癌 | 1/1 | 100% |
| 小細胞肺癌 | 0/1 | 0% |
安全性評価対象53例に認められた主な副作用は、ALT増加(35.8%)、AST増加(28.3%)、口内乾燥(26.4%)、下痢(17.0%)、高血圧(17.0%)等であった。(2023年1月13日データカットオフ)
17.3 その他
18. 薬効薬理
18.1 作用機序
セルペルカチニブは、RET、血管内皮増殖因子受容体(VEGFR)、線維芽細胞増殖因子受容体(FGFR)等のキナーゼ活性を阻害する。セルペルカチニブは、RET融合タンパク等のリン酸化を阻害し、下流のシグナル伝達分子のリン酸化を阻害することにより、腫瘍増殖抑制作用を示すと考えられている19)。
18.2 抗腫瘍効果
セルペルカチニブは、in vitroにおいて、RET融合タンパクを発現するヒト非小細胞肺癌由来LC-2/ad細胞株及びヒト甲状腺乳頭癌由来TPC-1細胞株並びに変異型RET(C634W及びM918T)をそれぞれ発現するヒト甲状腺髄様癌由来TT及びMZ-CRC1細胞株に対して増殖抑制作用を示した。また、セルペルカチニブは、in vivoにおいて、LC-2/ad細胞株、RET融合タンパクを発現する非小細胞肺癌患者由来CTG-0838腫瘍組織片及びTT細胞株をそれぞれ皮下移植した重症複合型免疫不全−ベージュマウス又はヌードマウスにおいて、腫瘍増殖抑制作用を示した19)。
19. 有効成分に関する理化学的知見
19.1. セルペルカチニブ
| 一般的名称 | セルペルカチニブ |
|---|---|
| 一般的名称(欧名) | Selpercatinib |
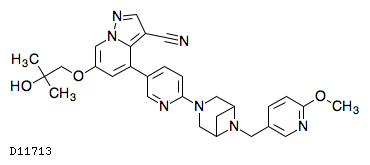
| 化学名 | 6-(2-Hydroxy-2-methylpropoxy)-4-(6-{6-[(6-methoxypyridin-3-yl)methyl]-3,6-diazabicyclo[3.1.1]heptan-3-yl}pyridin-3-yl)pyrazolo[1,5-a]pyridine-3-carbonitrile |
| 分子式 | C29H31N7O3 |
| 分子量 | 525.60 |
| 融点 | 約208℃ |
| 物理化学的性状 | 白色〜淡黄色の粉末である。0.1mol/L塩酸にやや溶けにくく、アセトン及びエタノールに極めて溶けにくく、水にほとんど溶けない。 |
| KEGG DRUG |
|
21. 承認条件
21.1 医薬品リスク管理計画を策定の上、適切に実施すること。
<RET融合遺伝子陽性の進行・再発の甲状腺癌、RET遺伝子変異陽性の根治切除不能な甲状腺髄様癌>
21.2 国内での治験症例が極めて限られていることから、製造販売後、一定数の症例に係るデータが集積されるまでの間は、全症例を対象に使用成績調査を実施することにより、本剤の使用患者の背景情報を把握するとともに、本剤の安全性及び有効性に関するデータを早期に収集し、本剤の適正使用に必要な措置を講じること。
22. 包装
<レットヴィモカプセル40mg>
56カプセル[8カプセル(PTP)×7]
<レットヴィモカプセル80mg>
56カプセル[8カプセル(PTP)×7]
23. 主要文献
- 社内資料:セルペルカチニブの毒性試験(2022年2月25日承認、CTD2.6.6)
- 社内資料:セルペルカチニブのがん原性試験
- 社内資料:進行癌患者を対象としたセルペルカチニブの第I/II相試験(LOXO-RET-17001試験)(2021年9月27日承認、CTD2.7.2.4.2、2.7.6.12、審査報告書)(2022年2月25日承認、審査報告書)
- 社内資料:セルペルカチニブの絶対的バイオアベイラビリティ及びマスバランス試験(LOXO-RET-18016試験)(2021年9月27日承認、CTD2.7.6.3)
- 社内資料:セルペルカチニブの薬物動態に及ぼす食事の影響及びオメプラゾールとの相互作用(LOXO-RET-18015試験)(2021年9月27日承認、CTD2.7.6.2)
- 社内資料:セルペルカチニブのヒト血漿蛋白結合率(LOXO-292-DMPK-060試験)(2021年9月27日承認、CTD2.7.2.3.2)
- 社内資料:セルペルカチニブのヒト血液中/血漿中濃度比(LOXO-292-DMPK-013試験)(2021年9月27日承認、CTD2.7.2.3.3)
- 社内資料:セルペルカチニブのCYP代謝(LOXO-292-DMPK-017試験)(2021年9月27日承認、CTD2.7.2.3.5)
- 社内資料:様々な重症度の腎機能障害を有する被験者におけるセルペルカチニブの薬物動態(LOXO-RET-18023試験)(2021年9月27日承認、CTD2.7.6.6)
- 社内資料:様々な重症度の肝機能障害を有する被験者におけるセルペルカチニブの薬物動態(LOXO-RET-18022試験)(2021年9月27日承認、CTD2.7.6.5)
- 社内資料:セルペルカチニブの母集団薬物動態解析(2022年2月25日承認、審査報告書)
- 社内資料:セルペルカチニブとイトラコナゾール及びリファンピシンの相互作用(LOXO-RET-18014試験)(2021年9月27日承認、CTD2.7.6.7)
- 社内資料:セルペルカチニブの生理学的薬物動態モデル解析(LOXO-292-DMPK-052試験)(2021年9月27日承認、CTD2.7.2.2.12)
- 社内資料:セルペルカチニブとミダゾラムの相互作用(LOXO-RET-18017試験)(2021年9月27日承認、CTD2.7.6.8)
- 社内資料:セルペルカチニブとレパグリニドの相互作用(LOXO-RET-18026試験)(2021年9月27日承認、CTD2.7.6.9)
- 社内資料:セルペルカチニブとラニチジン及びオメプラゾールの相互作用(LOXO-RET-19075試験)(2021年9月27日承認、CTD2.7.6.10)
- 社内資料:セルペルカチニブによるトランスポータ阻害(LOXO-292-DMPK-035試験)(2021年9月27日承認、CTD2.7.2.3.8)
- 社内資料:セルペルカチニブのQT/QTc評価(LOXO-RET-18032試験)(2021年9月27日承認、CTD2.7.2.2.8)
- 社内資料:セルペルカチニブの薬理試験(2021年9月27日承認、CTD2.6.2)
24. 文献請求先及び問い合わせ先
文献請求先
日本イーライリリー株式会社
医薬情報問合せ窓口
〒651-0086
神戸市中央区磯上通5丁目1番28号
電話:0120-360-605(医療関係者向け)
製品情報問い合わせ先
日本イーライリリー株式会社
医薬情報問合せ窓口
〒651-0086
神戸市中央区磯上通5丁目1番28号
電話:0120-360-605(医療関係者向け)
26. 製造販売業者等
26.1 製造販売元
日本イーライリリー株式会社
神戸市中央区磯上通5丁目1番28号