医薬品情報
| 総称名 | オゼンピック |
|---|---|
| 一般名 | セマグルチド(遺伝子組換え) |
| 欧文一般名 | Semaglutide(Genetical Recombination) |
| 製剤名 | セマグルチド(遺伝子組換え) |
| 薬効分類名 | 2型糖尿病治療剤 持続性GLP-1受容体作動薬 |
| 薬効分類番号 | 2499 |
| ATCコード | A10BJ06 |
| KEGG DRUG |
D10025
セマグルチド
|
| KEGG DGROUP |
DG01493
GLP-1受容体作動薬
DG02044
血糖降下薬
|
| JAPIC | 添付文書(PDF) |
添付文書情報2025年7月 改訂(第5版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| オゼンピック皮下注2mg | Ozempic Subcutaneous Injection 2mg | ノボノルディスクファーマ | 2499418G4027 | 11151円/キット | 劇薬, 処方箋医薬品注) |
2. 禁忌
次の患者には投与しないこと
2.1 本剤の成分に対し過敏症の既往歴のある患者
2.2 糖尿病性ケトアシドーシス、糖尿病性昏睡又は前昏睡、1型糖尿病の患者[インスリン製剤による速やかな治療が必須となるので、本剤を投与すべきでない。]
2.3 重症感染症、手術等の緊急の場合[インスリン製剤による血糖管理が望まれるので、本剤の投与は適さない。]
4. 効能または効果
5. 効能または効果に関連する注意
本剤の適用は、あらかじめ糖尿病治療の基本である食事療法、運動療法を十分に行ったうえで効果が不十分な場合に限り考慮すること。
6. 用法及び用量
通常、成人には、セマグルチド(遺伝子組換え)として週1回0.5mgを維持用量とし、皮下注射する。ただし、週1回0.25mgから開始し、4週間投与した後、週1回0.5mgに増量する。なお、患者の状態に応じて適宜増減するが、週1回0.5mgを4週間以上投与しても効果不十分な場合には、週1回1.0mgまで増量することができる。
7. 用法及び用量に関連する注意
7.1 本剤は週1回投与する薬剤であり、同一曜日に投与させること。
7.2 投与を忘れた場合は、次回投与までの期間が2日間(48時間)以上であれば、気づいた時点で直ちに投与し、その後はあらかじめ定めた曜日に投与すること。次回投与までの期間が2日間(48時間)未満であれば投与せず、次のあらかじめ定めた曜日に投与すること。なお、週1回投与の定めた曜日を変更する必要がある場合は、前回投与から少なくとも2日間(48時間)以上間隔を空けること。
8. 重要な基本的注意
8.1 本剤はインスリンの代替薬ではない。本剤の投与に際しては、患者のインスリン依存状態を確認し、投与の可否を判断すること。インスリン依存状態の患者で、インスリンからGLP-1受容体作動薬に切り替え、急激な高血糖及び糖尿病性ケトアシドーシスが発現した症例が報告されている。
8.2 投与する場合には、血糖、尿糖を定期的に検査し、薬剤の効果を確かめ、3〜4ヵ月間投与して効果が不十分な場合には、速やかに他の治療薬への切り替えを行うこと。
8.3 本剤は持続性製剤であり、本剤中止後も効果が持続する可能性があるため、血糖値の変動や副作用予防、副作用発現時の処置について十分留意すること。[16.1参照]
8.5 低血糖症状を起こすことがあるので、高所作業、自動車の運転等に従事している患者に投与するときには注意すること。[11.1.1参照]
8.6 急激な血糖コントロールの改善に伴い、糖尿病網膜症の顕在化又は増悪があらわれることがあるので、注意すること。
8.9 下痢、嘔吐から脱水を続発し、急性腎障害に至るおそれがあるので、患者の状態に注意すること
8.10 本剤投与中は、甲状腺関連の症候の有無を確認し、異常が認められた場合には、専門医を受診するよう指導すること。[15.2参照]
8.11 胆石症、胆嚢炎、胆管炎又は胆汁うっ滞性黄疸が発現するおそれがあるので、腹痛等の腹部症状がみられた場合には、必要に応じて画像検査等による原因精査を考慮するなど、適切に対応すること。[11.1.3参照]
8.12 本剤の自己注射にあたっては、以下の点に留意すること。
・投与法について十分な教育訓練を実施したのち、患者自ら確実に投与できることを確認した上で、医師の管理指導の下で実施すること。
・全ての器具の安全な廃棄方法について指導を徹底すること。
・添付されている取扱説明書を必ず読むよう指導すること。
8.13 本剤はセマグルチド(遺伝子組換え)を含有しているため、ウゴービ等他のセマグルチド(遺伝子組換え)含有製剤と併用しないこと。
8.14 本剤とDPP-4阻害剤はいずれもGLP-1受容体を介した血糖降下作用を有している。両剤を併用した際の臨床試験成績はなく、有効性及び安全性は確認されていない。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.2 重度胃不全麻痺等、重度の胃腸障害のある患者
十分な使用経験がなく、胃腸障害の症状が悪化するおそれがある。
9.1.3 低血糖を起こすおそれがある以下の患者又は状態
9.1.4 腹部手術の既往又はイレウスの既往のある患者
腸閉塞を含むイレウスを起こすおそれがある。[11.1.4参照]
9.4 生殖能を有する者
2ヵ月以内に妊娠を予定する女性には本剤を投与せず、インスリンを使用すること。[9.5参照]
9.5 妊婦
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。
ラットで乳汁中への移行が報告されている。ヒトでの乳汁移行に関するデータ及びヒトの哺乳中の児への影響に関するデータはない。
ラットで乳汁中への移行が報告されている。ヒトでの乳汁移行に関するデータ及びヒトの哺乳中の児への影響に関するデータはない。
9.7 小児等
小児等を対象とした臨床試験は実施していない。
9.8 高齢者
患者の状態を観察しながら慎重に投与すること。一般に生理機能が低下していることが多い。[16.6.3参照]
10. 相互作用
10.2 併用注意
| 糖尿病用薬 ビグアナイド系薬剤 スルホニルウレア剤 速効型インスリン分泌促進剤 α-グルコシダーゼ阻害剤 チアゾリジン系薬剤 DPP-4阻害剤 SGLT2阻害剤 インスリン製剤 等 [11.1.1参照] | 低血糖症の発現に注意すること。特に、インスリン製剤又はスルホニルウレア剤と併用する場合、低血糖のリスクが増加するおそれがあるため、定期的な血糖測定を行い、必要に応じ、これらの薬剤の減量を検討すること。 | 血糖降下作用が増強される。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 低血糖(頻度不明)
11.1.2 急性膵炎(頻度不明)
11.1.3 胆嚢炎、胆管炎、胆汁うっ滞性黄疸(いずれも頻度不明)[8.11参照]
11.1.4 イレウス(頻度不明)
腸閉塞を含むイレウスを起こすおそれがある。高度の便秘、腹部膨満、持続する腹痛、嘔吐等の異常が認められた場合には投与を中止し、適切な処置を行うこと。[9.1.4参照]
11.2 その他の副作用
| 5%以上 | 1〜5%未満 | 0.5〜1%未満 | 頻度不明 | |
| 感染症 | 胃腸炎 | |||
| 免疫系障害 | 過敏症(発疹、じん麻疹等) | |||
| 代謝及び栄養障害 | 食欲減退 | |||
| 神経系障害 | 頭痛 | 浮動性めまい | 味覚異常 | |
| 眼障害 | 糖尿病網膜症関連事象 | |||
| 心臓障害 | 心拍数増加注1 | |||
| 胃腸障害 | 悪心、下痢、便秘、嘔吐 | 腹部不快感、消化不良、腹部膨満、上腹部痛、腹痛、おくび | 胃食道逆流性疾患、鼓腸、胃炎 | 胃排出遅延 |
| 肝胆道系障害 | 胆石症 | |||
| 皮膚及び皮下組織 | 血管性浮腫 | |||
| 全身障害及び投与部位状態 | 疲労、無力症 | 注射部位反応 | ||
| 臨床検査注2 | リパーゼ増加 | アミラーゼ増加、体重減少 | 血中クレアチンホスホキナーゼ増加 |
14. 適用上の注意
14.1 薬剤投与時の注意
14.1.1 投与時
(1)本剤はJIS T 3226-2に準拠したA型専用注射針を用いて使用すること。本剤はA型専用注射針との適合性の確認をペンニードルで行っている。
(2)本剤とA型専用注射針との装着時に液漏れが認められた場合には、新しい注射針に取り替えること。
(3)1本の本剤を複数の患者に使用しないこと。
14.1.2 投与部位
皮下注射は、腹部、大腿、上腕に行う。注射箇所は毎回変更し、少なくとも前回の注射箇所より2〜3cm離すこと。
14.1.3 投与経路
静脈内及び筋肉内に投与しないこと。
14.1.4 その他
(1)本剤は他の製剤との混合により、成分が分解するおそれがあるため、本剤と他の製剤を混合しないこと。
(2)注射後は必ず注射針を外すこと。注射針は毎回新しいものを、必ず注射直前に取り付けること。
針を付けたままにすると、液漏れや針詰まりにより正常に注射できないおそれがある。また、薬剤の濃度変化や感染症の原因となることがある。
針を付けたままにすると、液漏れや針詰まりにより正常に注射できないおそれがある。また、薬剤の濃度変化や感染症の原因となることがある。
(3)カートリッジの内壁に付着物がみられたり、液中に塊や薄片がみられることがある。また、使用中に液が変色することがある。これらのような場合は使用しないこと。
15. その他の注意
15.2 非臨床試験に基づく情報
16. 薬物動態
16.1 血中濃度
健康男性被験者における反復皮下投与後の薬物動態
日本人健康男性被験者を対象に、本剤0.5mg(8例)及び1.0mg(8例)の13週間反復皮下投与後の薬物動態プロファイルを検討した。本剤は、週1回0.25mgで投与を開始し、4週間投与した後に週1回0.5mgへ増量した。1.0mgまで増量する群では、その後週1回0.5mgを4週間投与した後に週1回1.0mgへ増量した。
定常状態での血漿中濃度−時間推移及び薬物動態パラメータを以下に示す7)。[8.3参照]
定常状態での血漿中濃度−時間推移及び薬物動態パラメータを以下に示す7)。[8.3参照]
| 用量 | N | AUC0-168h(nmol・h/L) | Cmax(nmol/L) | tmax注)(h) | t1/2(h) | CL/F(L/h) | Vz/F(L) |
| 0.5mg | 8 | 3583(17.8) | 25.1(17.8) | 30(12-72) | 145(8.0) | 0.034(17.8) | 7.1(12.8) |
| 1.0mg | 8 | 7449(12.2) | 51.6(11.1) | 36(18-96) | 163(10.9) | 0.033(12.2) | 7.7(14.0) |
16.2 吸収
外国人健康成人10例に本剤0.5mgを単回皮下投与したときの絶対的バイオアベイラビリティは、89%であった8)。
2型糖尿病患者1612例(うち日本人555例)を対象とした母集団薬物動態解析の結果、本剤を異なる投与部位(腹部、大腿部及び上腕部)に投与したとき、腹部への投与に対する大腿部及び上腕部への投与での定常状態の本剤曝露量の比の推定値及び90%信頼区間は、0.96[0.93;1.00]及び0.92[0.89;0.96]であった。
16.3 分布
16.4 代謝
16.5 排泄
16.6 特定の背景を有する患者
16.6.1 腎機能障害被験者における薬物動態
腎機能障害の程度の異なる被験者(クレアチニンクリアランス(Ccr)による分類)における本剤0.5mg単回皮下投与後の薬物動態を、腎機能が正常な被験者(Ccr 80mL/min超)と比較検討した結果を以下に示す9)(外国人データ)。
| 腎機能 | AUC0-inf | Cmax |
| 比の推定値 [95%信頼区間] | 比の推定値 [90%信頼区間] | |
| 軽度/正常 (軽度:Ccr50超〜80mL/min) | 0.99 [0.85;1.16] | 0.90 [0.73;1.11] |
| 中等度/正常 (中等度:Ccr30超〜50mL/min) | 1.07 [0.91;1.27] | 0.79 [0.64;0.99] |
| 重度/正常 (重度:Ccr30mL/min以下) | 1.13 [0.97;1.32] | 0.86 [0.70;1.06] |
| 末期/正常 (末期:血液透析を必要とする被験者) | 1.10 [0.94;1.28] | 0.82 [0.66;1.01] |
16.6.2 肝機能障害被験者における薬物動態
肝機能障害の程度の異なる被験者(Child-Pugh scoresに基づく分類)における本剤0.5mg単回皮下投与後の薬物動態を、肝機能が正常な被験者と比較検討した結果を以下に示す10)(外国人データ)。
| 肝機能 | AUC0-inf | Cmax |
| 比の推定値 [90%信頼区間] | 比の推定値 [90%信頼区間] | |
| 軽度/正常 (軽度:Child-Pugh分類A) | 0.95 [0.77;1.16] | 0.99 [0.80;1.23] |
| 中等度/正常 (中等度:Child-Pugh分類B) | 1.02 [0.93;1.12] | 1.02 [0.88;1.18] |
| 重度/正常 (重度:Child-Pugh分類C) | 0.97 [0.84;1.12] | 1.15 [0.89;1.48] |
16.6.3 高齢者における薬物動態
2型糖尿病患者1612例(うち日本人555例)を対象とした母集団薬物動態解析の結果、65歳未満に対する65歳以上〜75歳未満及び75歳以上の定常状態の平均血漿中濃度の比と90%信頼区間は1.01[0.99;1.03]及び1.04[1.00;1.09]と推定された。[9.8参照]
16.7 薬物相互作用
本剤1.0mgの定常状態において、メトホルミン、ワルファリン、ジゴキシン、アトルバスタチン、経口避妊薬及びアセトアミノフェンを併用投与したときの薬物動態の結果を以下に示す15)16)17)(外国人データ)。[18.2.4参照]
| 経口薬 | 用量amg | 対象 | N | AUCb比c [90%信頼区間]e | Cmax比c [90%信頼区間]e | tmax差d [90%信頼区間] |
| メトホルミン | 500 | 健康被験者 | 22 | 1.03 [0.96;1.11] | 0.90 [0.83;0.98] | 0.50 [−0.38;1.25] |
| S-ワルファリン | 25 | 健康被験者 | 22 | 1.05 [0.99;1.11] | 0.91 [0.85;0.98] | 2.00 [1.25;2.75] |
| R-ワルファリン | 25 | 健康被験者 | 22 | 1.04 [0.98;1.10] | 0.93 [0.87;1.00] | 1.75 [0.88;2.50] |
| ジゴキシン | 0.5 | 健康被験者 | 26 | 1.02 [0.97;1.08] | 0.93 [0.84;1.03] | 0.25 [0.00;0.25] |
| アトルバスタチン | 40 | 健康被験者 | 26 | 1.02 [0.93;1.12] | 0.62 [0.47;0.82] | 1.75 [1.00;2.50] |
| エチニルエストラジオール | 0.03 | 2型糖尿病 | 37 | 1.11 [1.06;1.15] | 1.04 [0.98;1.10] | 0.50 [0.00;0.50] |
| レボノルゲストレル | 0.15 | 2型糖尿病 | 40 | 1.20 [1.15;1.26] | 1.05 [0.99;1.12] | 0.50 [0.25;0.75] |
| パラセタモール (アセトアミノフェン) | 1500 | 肥満被験者 | 28 | 0.94 [0.88;1.01] | 0.77 [0.67;0.88] | 0.25 [0.13;0.25] |
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 単独療法:プラセボ対照二重盲検比較試験(第III相国際共同試験)
食事療法及び運動療法で血糖コントロールが不十分な2型糖尿病患者388例を対象に無作為割り付けを行い、二重盲検下で本剤0.5mg、本剤1.0mg又はプラセボを週1回、30週間投与した(本剤0.5mg群:128例(日本人:19例)、本剤1.0mg群:130例(日本人:19例)、プラセボ群:129例(日本人:23例))。本剤は、週1回0.25mgで投与を開始し、4週間投与した後に週1回0.5mgへ増量した。1.0mgまで増量する群では、その後週1回0.5mgを4週間投与した後に週1回1.0mgへ増量した。
本剤0.5mg及び1.0mgの30週間投与により、主要評価項目であるHbA1cのベースラインから投与後30週までの変化量に関して、本剤のいずれの用量でもプラセボに対する優越性が検証された(p<0.0001、下表参照)。
本剤0.5mg及び1.0mgの30週間投与により、主要評価項目であるHbA1cのベースラインから投与後30週までの変化量に関して、本剤のいずれの用量でもプラセボに対する優越性が検証された(p<0.0001、下表参照)。
| HbA1c(%) | 本剤0.5mg | 本剤1.0mg | プラセボ |
| ベースラインa | 8.09±0.89(128) | 8.12±0.81(130) | 7.95±0.85(129) |
| 投与後30週までの変化量a | −1.56±1.02(102) | −1.73±1.15(104) | −0.15±0.94(84) |
| 群差(本剤−プラセボ)b [95%信頼区間] | −1.43 [−1.71;−1.15] | −1.53 [−1.81;−1.25] | − |
ベースラインから投与後30週までの体重の変化量(最小二乗平均±標準誤差)は、本剤0.5mg群で−3.7±0.41kg(ベースラインの平均:89.8kg)、本剤1.0mg群で−4.5±0.41kg(ベースラインの平均:96.9kg)、プラセボ群で−1.0±0.43kg(ベースラインの平均:89.1kg)であった。
重大な低血糖は報告されなかった。重大な又は血糖値確定(56mg/dL未満)症候性低血糖注)は本剤群ではいずれの用量でも認められなかったが、プラセボ群では2例3件報告された18)。[11.1.1参照]
重大な低血糖は報告されなかった。重大な又は血糖値確定(56mg/dL未満)症候性低血糖注)は本剤群ではいずれの用量でも認められなかったが、プラセボ群では2例3件報告された18)。[11.1.1参照]
注)重大な低血糖(米国糖尿病学会分類による)又は低血糖症状を伴う血糖値(血漿)が56mg/dL未満の低血糖。
17.1.2 併用療法:メトホルミン又はチアゾリジン系薬剤あるいは両剤との併用、実薬対照二重盲検比較試験(第III相国際共同試験)
2型糖尿病患者1231例を対象に無作為割り付けを行い、メトホルミン又はチアゾリジン系薬剤あるいはこれら2剤による併用療法に追加して、二重盲検下で本剤0.5mg又は本剤1.0mgを週1回、あるいはシタグリプチン100mgを1日1回、56週間投与した(本剤0.5mg群:409例(日本人:48例)、本剤1.0mg群:409例(日本人:43例)、シタグリプチン群:407例(日本人:49例))。本剤は、週1回0.25mgで投与を開始し、4週間投与した後に週1回0.5mgへ増量した。1.0mgまで増量する群では、その後週1回0.5mgを4週間投与した後に週1回1.0mgへ増量した。
主要評価項目であるHbA1cのベースラインから投与後56週までの変化量に関して、本剤0.5mg及び本剤1.0mgのシタグリプチンに対する非劣性が検証された(非劣性マージン:0.3%)(下表参照)。
主要評価項目であるHbA1cのベースラインから投与後56週までの変化量に関して、本剤0.5mg及び本剤1.0mgのシタグリプチンに対する非劣性が検証された(非劣性マージン:0.3%)(下表参照)。
| HbA1c(%) | 本剤0.5mg | 本剤1.0mg | シタグリプチン100mg |
| ベースラインa | 8.01±0.92(409) | 8.04±0.93(409) | 8.17±0.92(407) |
| 投与後56週までの変化量a | −1.40±1.08(328) | −1.64±1.04(331) | −0.79±1.05(285) |
| 群差(本剤−シタグリプチン)b [95%信頼区間] | −0.77 [−0.92;−0.62] | −1.06 [−1.21;−0.91] | − |
日本人でのメトホルミン単剤との併用結果を下表に示す。
| HbA1c(%) | 本剤0.5mg | 本剤1.0mg | シタグリプチン100mg |
| ベースラインa | 8.16±0.97(46) | 8.06±0.96(42) | 8.41±0.80(48) |
| 投与後56週までの変化量a | −1.84±0.81(41) | −1.97±0.80(34) | −0.82±1.02(35) |
| 群差(本剤−シタグリプチン)b [95%信頼区間] | −1.29 [−1.64;−0.93] | −1.52 [−1.90;−1.15] | − |
17.1.3 併用療法:Basalインスリンとの併用、プラセボ対照二重盲検比較試験(第III相国際共同試験)
Basalインスリンの単独療法又はBasalインスリンとメトホルミンとの併用療法で血糖コントロールが不十分な2型糖尿病患者397例を対象に無作為割り付けを行い、二重盲検下で本剤0.5mg、本剤1.0mg又はプラセボを週1回、30週間追加投与した(本剤0.5mg群:132例(日本人:17例)、本剤1.0mg群:131例(日本人:22例)、プラセボ群:133例(日本人:22例))。本剤は、週1回0.25mgで投与を開始し、4週間投与した後に週1回0.5mgへ増量した。1.0mgまで増量する群では、その後週1回0.5mgを4週間投与した後に週1回1.0mgへ増量した。なお、スクリーニング時のHbA1cが8.0%以下の場合には、低血糖のリスクを低減するために併用投与開始時のインスリン用量を20%減量した。
本剤0.5mg及び1.0mgの30週間投与により、主要評価項目であるHbA1cのベースラインから投与後30週までの変化量に関して、本剤のいずれの用量でもプラセボに対する優越性が検証された(p<0.0001、下表参照)。
本剤0.5mg及び1.0mgの30週間投与により、主要評価項目であるHbA1cのベースラインから投与後30週までの変化量に関して、本剤のいずれの用量でもプラセボに対する優越性が検証された(p<0.0001、下表参照)。
| HbA1c(%) | 本剤0.5mg | 本剤1.0mg | プラセボ |
| ベースラインa | 8.36±0.83(132) | 8.31±0.82(131) | 8.42±0.88(133) |
| 投与後30週までの変化量a | −1.46±1.08(111) | −1.87±0.91(108) | −0.19±1.07(94) |
| 群差(本剤−プラセボ)b [95%信頼区間] | −1.35 [−1.61;−1.10] | −1.75 [−2.01;−1.50] | − |
17.1.4 単独療法:実薬対照非盲検比較試験(第III相国内試験)
経口糖尿病薬の単独療法又は食事療法及び運動療法で治療中の日本人2型糖尿病患者308例を対象に無作為割り付けを行い、本剤0.5mg又は本剤1.0mgを週1回、あるいはシタグリプチン100mgを1日1回、30週間投与した(本剤0.5mg群:103例、本剤1.0mg群:102例、シタグリプチン群:103例)。本剤は、週1回0.25mgで投与を開始し、4週間投与した後に週1回0.5mgへ増量した。1.0mgまで増量する群では、その後週1回0.5mgを4週間投与した後に週1回1.0mgへ増量した。
本試験の結果を下表に示す。
本試験の結果を下表に示す。
| HbA1c(%) | 本剤0.5mg | 本剤1.0mg | シタグリプチン100mg |
| ベースラインa | 8.23±1.02(103) | 8.01±0.85(102) | 8.20±0.89(103) |
| 投与後30週までの変化量a | −1.93±0.97(98) | −2.14±1.00(87) | −0.83±0.82(95) |
| 群差(本剤−シタグリプチン)b [95%信頼区間] | −1.13 [−1.32;−0.94] | −1.44 [−1.63;−1.24] | − |
17.1.5 非盲検長期(56週間)安全性試験(第III相国内試験)
経口糖尿病薬の単独療法又は食事療法及び運動療法で血糖コントロールが不十分な日本人2型糖尿病患者601例を対象に無作為割り付けを行い、本剤0.5mg又は本剤1.0mgを週1回(単独療法あるいは経口糖尿病薬単剤(スルホニルウレア剤、速効型インスリン分泌促進剤、α-グルコシダーゼ阻害剤又はチアゾリジン系薬剤のいずれか)との併用療法)、あるいは追加の経口糖尿病薬(前治療と異なる機序による薬剤;国内で承認された効能又は効果、用法及び用量に従う)を、56週間追加投与した(本剤0.5mg群:239例、本剤1.0mg群:241例、追加の経口糖尿病薬群:120例)。本剤は、週1回0.25mgで投与を開始し、4週間投与した後に週1回0.5mgへ増量した。1.0mgまで増量する群では、その後週1回0.5mgを4週間投与した後に週1回1.0mgへ増量した。
本剤群における結果を下表に示す。
本剤群における結果を下表に示す。
| HbA1c(%) | ベースライン | 投与後56週までの変化量 | |
| 本剤0.5mg | |||
| 単独療法 | 7.86±0.78(68) | −1.77±0.87(64) | |
| スルホニルウレア剤 | 8.49±0.92(68) | −1.85±0.89(64) | |
| 速効型インスリン分泌促進剤 | 7.77±0.58(34) | −1.48±0.90(31) | |
| α-グルコシダーゼ阻害剤 | 8.23±1.10(35) | −2.13±1.06(32) | |
| チアゾリジン系薬剤 | 7.60±0.62(34) | −1.27±0.89(29) | |
| 本剤1.0mg | |||
| 単独療法 | 7.94±0.84(68) | −1.99±0.83(53) | |
| スルホニルウレア剤 | 8.23±0.96(69) | −2.17±0.97(59) | |
| 速効型インスリン分泌促進剤 | 8.49±0.85(36) | −2.33±0.90(33) | |
| α-グルコシダーゼ阻害剤 | 7.92±0.75(34) | −2.04±0.80(29) | |
| チアゾリジン系薬剤 | 8.22±1.34(34) | −2.08±1.28(30) | |
18. 薬効薬理
18.1 作用機序
本剤はヒトGLP-1アナログであり、内因性GLP-1が標的とするGLP-1受容体と選択的に結合し、cAMP放出量を増加させるGLP-1受容体作動薬として作用する。
本剤はアルブミンと結合して代謝による分解の遅延及び腎クリアランスの低下を示すと考えられており、またアミノ酸置換によりDPP-4による分解に対して抵抗性を示すことにより、作用が持続する。
本剤はアルブミンと結合して代謝による分解の遅延及び腎クリアランスの低下を示すと考えられており、またアミノ酸置換によりDPP-4による分解に対して抵抗性を示すことにより、作用が持続する。
18.2 薬理作用
ヒトでの薬力学的作用の評価は、特記する場合を除き、すべて本剤1.0mgの週1回12週間(用量漸増期間を含む)皮下投与後の定常状態において行われた。
18.2.1 血糖降下作用
本剤の投与により、糖尿病db/dbマウス(1日1回28日間反復投与)で溶媒対照群と比較し血糖値が低下した23)。
外国人2型糖尿病患者において、本剤の投与によりグルコース濃度依存的にインスリン分泌が促進及びグルカゴン分泌が抑制され、血中グルコース濃度はプラセボと比較して低下した24)。
外国人2型糖尿病患者に本剤1.0mgを週1回13週間(用量漸増期間を含む)皮下投与した結果、最終投与後1週間における空腹時血糖値はプラセボと比較して低く、血糖降下作用は1週間後においても持続していた25)。
18.2.2 グルコース応答性インスリン分泌
外国人2型糖尿病患者に本剤を投与した結果、静脈内グルコース急速注入後のインスリンの第1相分泌(グルコース投与直後から10分後)及び第2相分泌(グルコース投与10分後から120分後)反応は、プラセボと比較して増加した24)。
18.2.3 グルカゴン分泌
外国人2型糖尿病患者において、本剤投与により、プラセボと比較して空腹時グルカゴン濃度及び食後のグルカゴン分泌反応が低下した24)。
18.2.4 胃内容排出
19. 有効成分に関する理化学的知見
19.1. セマグルチド(遺伝子組換え)
| 一般的名称 | セマグルチド(遺伝子組換え) |
|---|---|
| 一般的名称(欧名) | Semaglutide(Genetical Recombination) |
| 分子式 | C187H291N45O59 |
| 分子量 | 4113.58 |
| 理化学知見その他 | セマグルチドは、遺伝子組換えヒトグルカゴン様ペプチド-1(GLP-1)類縁体であり、ヒトGLP-1の7〜37番目のアミノ酸に相当し、2番目のAla及び28番目のLysは、それぞれ2-アミノ-2-メチルプロパン酸及びArgに置換され、1,18-オクタデカン二酸が1個のGlu及び2個の8-アミノ-3,6-ジオキサオクタン酸で構成されるリンカーを介して20番目のLysに結合している。セマグルチドは、31個のアミノ酸残基からなる修飾ペプチドである。 |
| KEGG DRUG |
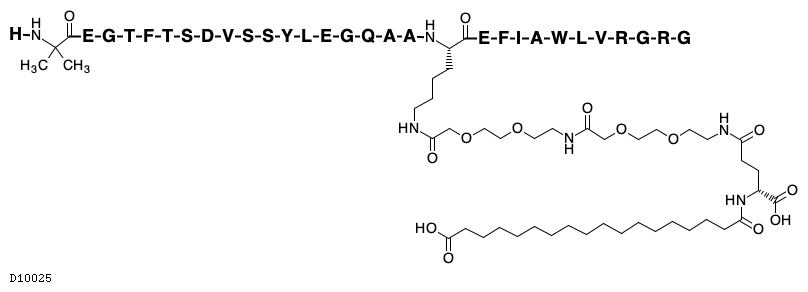
|
20. 取扱い上の注意
使用開始後は遮光にて室温(冷蔵庫(2〜8℃)も含む)に保管し、8週間以内に使用すること。
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
1筒1.5mL
2本
23. 主要文献
- 社内資料:ラットを用いた受胎能及びEFD試験(2018年3月23日承認,CTD2.6.6.6.1)
- 社内資料:ウサギを用いたEFD試験(2018年3月23日承認,CTD2.6.6.6.2)
- 社内資料:カニクイザルを用いたEFD試験(2018年3月23日承認,CTD2.6.6.6.3)
- 社内資料:カニクイザルを用いたEFD及びPPND試験(2018年3月23日承認,CTD2.6.6.6.3)
- 社内資料:ラットを用いた104週間反復皮下投与がん原性試験(2018年3月23日承認,CTD2.6.6.5)
- 社内資料:マウスを用いた104週間反復皮下投与がん原性試験(2018年3月23日承認,CTD2.6.6.5)
- Ikushima I.,et al., Adv Ther., 35 (4), 531-44, (2018) »PubMed
- 社内資料:第I相臨床試験(NN9535-3687)(2018年3月23日承認,CTD2.7.2.3)
- Marbury T.C.,et al., Clin Pharmacokinet., 56 (11), 1381-90, (2017) »PubMed
- Jensen L.,et al., Diabetes Obes Metab., 20 (4), 998-1005, (2018) »PubMed
- Jensen L.,et al., Eur J Pharm Sci., 104, 31-41, (2017) »PubMed
- 社内資料:酵素誘導(2018年3月23日承認,CTD2.6.4.7)
- 社内資料:酵素阻害(2018年3月23日承認,CTD2.6.4.7)
- 社内資料:トランスポーター阻害(2018年3月23日承認,CTD2.6.4.7)
- Kapitza C.,et al., J Clin Pharmacol., 55 (5), 497-504, (2015) »PubMed
- Hausner H.,et al., Clin Pharmacokinet., 56 (11), 1391-401, (2017) »PubMed
- Blundell J.,et al., Diabetes Obes Metab., 19 (9), 1242-51, (2017) »PubMed
- Sorli C.,et al., Lancet Diabetes Endocrinol., 5 (4), 251-60, (2017) »PubMed
- Ahren B.,et al., Lancet Diabetes Endocrinol., 5 (5), 341-54, (2017) »PubMed
- Rodbard HW.,et al., J Clin Endocrinol Metab., 103 (6), 2291-301, (2018) »PubMed
- Seino Y.,et al., Diabetes Obes Metab., 20 (2), 378-88, (2018) »PubMed
- Kaku K.,et al., Diabetes Obes Metab., 20 (5), 1202-12, (2018) »PubMed
- 社内資料:db/dbマウスにおける体重、摂餌量、血糖及びβ細胞容積ならびに機能への影響(2018年3月23日承認,CTD2.6.2.2.2.3)
- Kapitza C.,et al., Diabetologia., 60 (8), 1390-9, (2017) »PubMed
- 社内資料:第I相臨床試験(NN9535-3684)(2018年3月23日承認,CTD2.5.3.4)
- 社内資料:摘出灌流ラット膵臓からのインスリン分泌(2018年3月23日承認,CTD2.6.2.2.1.2)
- 社内資料:ミニブタのインスリン分泌に及ぼす薬理作用持続時間の検討(2018年3月23日承認,CTD2.6.2.2.2.4)
24. 文献請求先及び問い合わせ先
文献請求先
ノボノルディスクファーマ株式会社
ノボケア相談室
〒100-0005
東京都千代田区丸の内2-1-1
電話:0120-180363(フリーダイアル)
住友ファーマ株式会社
くすり情報センター
〒541-0045
大阪市中央区道修町2-6-8
電話:0120-034-389
製品情報問い合わせ先
ノボノルディスクファーマ株式会社
ノボケア相談室
〒100-0005
東京都千代田区丸の内2-1-1
電話:0120-180363(フリーダイアル)
住友ファーマ株式会社
くすり情報センター
〒541-0045
大阪市中央区道修町2-6-8
電話:0120-034-389
26. 製造販売業者等
26.1 製造販売元
26.2 プロモーション提携
住友ファーマ株式会社
大阪市中央区道修町2-6-8