医薬品情報
| 総称名 | ボカブリア |
|---|---|
| 一般名 | カボテグラビル |
| 欧文一般名 | Cabotegravir |
| 製剤名 | カボテグラビル持効性懸濁注射液 |
| 薬効分類名 | 持効性HIVインテグラーゼ阻害剤 |
| 薬効分類番号 | 6250 |
| ATCコード | J05AJ04 |
| KEGG DRUG |
D10548
カボテグラビル
|
| KEGG DGROUP |
DG01628
カボテグラビル
DG03107
抗HIV薬
|
| JAPIC | 添付文書(PDF) |
添付文書情報2024年1月 改訂(第4版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ボカブリア水懸筋注400mg | VOCABRIA Aqueous Suspension for IM Injection | ヴィーブヘルスケア | 6250408A1028 | 176458円/瓶 | 処方箋医薬品注) |
| ボカブリア水懸筋注600mg | VOCABRIA Aqueous Suspension for IM Injection | ヴィーブヘルスケア | 6250408A2024 | 253850円/瓶 | 処方箋医薬品注) |
2. 禁忌
次の患者には投与しないこと
2.1 本剤の成分に対し過敏症の既往歴のある患者
2.2 リファンピシン、フェニトイン、ホスフェニトイン、フェノバルビタール、カルバマゼピンを投与中の患者[10.1参照]
4. 効能または効果
5. 効能または効果に関連する注意
5.1 本剤は、ウイルス学的失敗の経験がなく、切り替え前6ヵ月間以上においてウイルス学的抑制(ヒト免疫不全ウイルス[HIV]-1 RNA量が50copies/mL未満)が得られており、カボテグラビル及びリルピビリンに対する耐性関連変異を持たず、本剤への切り替えが適切であると判断される抗HIV薬既治療患者に使用すること。[17.1.1-17.1.3参照]
5.2 本剤の投与の前にカボテグラビル経口剤をリルピビリン経口剤との併用により1ヵ月間(少なくとも28日間)を目安に経口投与し、カボテグラビル及びリルピビリンに対する忍容性が確認された患者を対象とすること。
5.3 本剤による治療にあたっては、患者の治療歴及び可能な場合には薬剤耐性検査(遺伝子型解析あるいは表現型解析)を参考にすること。
6. 用法及び用量
<1ヵ月間隔投与>
リルピビリンとの併用において、通常、成人にはカボテグラビルとして600mgを臀部筋肉内に投与する。以降は、400mgを1ヵ月に1回、臀部筋肉内に投与する。
<2ヵ月間隔投与>
リルピビリンとの併用において、通常、成人にはカボテグラビルとして600mgを臀部筋肉内に投与する。本剤初回投与1ヵ月後に600mgを臀部筋肉内に投与し、以降は600mgを2ヵ月に1回、臀部筋肉内に投与する。
7. 用法及び用量に関連する注意
<共通>
7.1 本剤の投与スケジュールを遵守すること。投与スケジュールを遵守できなかった場合は、本剤の継続の可否も含め、治療法を再考すること。
7.2 本剤の初回投与は、カボテグラビル経口剤及びリルピビリン経口剤の投与最終日と同日に行うこと。
7.3 本剤の用法及び用量は、患者の状態及びリスク・ベネフィットを考慮して、医師が慎重に選択すること。[17.1.3参照]
<1ヵ月間隔投与>
7.4 本剤の2回目以降の投与は、投与予定日の前後7日以内に投与すること。投与予定日の7日後までに投与できない場合は、代替としてカボテグラビル経口剤とリルピビリン経口剤を1日1回併用経口投与すること。経口剤による代替期間が2ヵ月間を超える場合は、他の抗HIV薬へ切り替えることを考慮すること。
7.5 代替経口投与後、本剤の1ヵ月間隔投与を再開する場合は、本剤最終投与からの期間に基づき、下表に従い再開すること。なお、本剤の投与再開は、代替経口投与最終日と同日に行うこと。
| 本剤最終投与からの期間 | 再開時の用法及び用量 |
| 2ヵ月以内 | 可能な限り早期にカボテグラビル400mgを1回臀部筋肉内に投与して再開する。再開以降はカボテグラビル400mgを1ヵ月に1回臀部筋肉内に投与する。 |
| 2ヵ月超 | カボテグラビル600mgを1回臀部筋肉内に投与して再開する。再開以降はカボテグラビル400mgを1ヵ月に1回臀部筋肉内に投与する。 |
7.6 1ヵ月間隔投与から2ヵ月間隔投与に切り替える際は、カボテグラビル400mgを最終投与した1ヵ月後に、カボテグラビル600mgを1回臀部筋肉内に投与し、以降はカボテグラビル600mgを2ヵ月に1回臀部筋肉内に投与すること。
<2ヵ月間隔投与>
7.7 本剤の2回目以降の投与は、投与予定日の前後7日以内に投与すること。投与予定日の7日後までに投与できない場合は、代替としてカボテグラビル経口剤とリルピビリン経口剤を1日1回併用経口投与すること。経口剤による代替期間が2ヵ月間を超える場合は、他の抗HIV薬へ切り替えることを考慮すること。
7.8 代替経口投与後、本剤の2ヵ月間隔投与を再開する場合は、本剤最終投与からの期間に基づき、下表に従い再開すること。なお、本剤の投与再開は、代替経口投与最終日と同日に行うこと。
| 経口投与により代替した本剤投与 | 本剤最終投与からの期間 | 再開時の用法及び用量 |
| 本剤2回目 | 2ヵ月以内 | 可能な限り早期にカボテグラビル600mgを1回臀部筋肉内に投与して再開する。再開以降はカボテグラビル600mgを2ヵ月に1回臀部筋肉内に投与する。 |
| 2ヵ月超 | カボテグラビル600mgを1回臀部筋肉内に投与して再開する。再開1ヵ月後にカボテグラビル600mgを1回臀部筋肉内に投与し、以降はカボテグラビル600mgを2ヵ月に1回臀部筋肉内に投与する。 | |
| 本剤3回目以降 | 3ヵ月以内 | 可能な限り早期にカボテグラビル600mgを1回臀部筋肉内に投与して再開する。再開以降はカボテグラビル600mgを2ヵ月に1回臀部筋肉内に投与する。 |
| 3ヵ月超 | カボテグラビル600mgを1回臀部筋肉内に投与して再開する。再開1ヵ月後にカボテグラビル600mgを1回臀部筋肉内に投与し、以降はカボテグラビル600mgを2ヵ月に1回臀部筋肉内に投与する。 |
7.9 2ヵ月間隔投与から1ヵ月間隔投与に切り替える際は、カボテグラビル600mgを最終投与した2ヵ月後に、カボテグラビル400mgを1回臀部筋肉内に投与し、以降はカボテグラビル400mgを1ヵ月に1回臀部筋肉内に投与すること。
8. 重要な基本的注意
8.1 本剤による治療は、抗HIV療法に十分な経験を持つ医師のもとで開始すること。
8.2 本剤は投与スケジュールが遵守されない場合、ウイルスの再増殖及び薬剤耐性リスクのおそれがあるため、投与スケジュールを遵守するよう患者に指導すること。
8.3 本剤の使用に際しては、国内外のガイドライン等の最新の情報を参考に、患者又は患者に代わる適切な者に、次の事項についてよく説明し同意を得た後、使用すること。
・本剤はHIV感染症の根治療法薬ではないことから、日和見感染を含むHIV感染症の進展に伴う疾病を発症し続ける可能性があるので、本剤投与開始後の身体状況の変化については、すべて担当医に報告すること。
・本剤の長期投与による影響については、現在のところ不明であること。
・本剤は併用薬剤と相互作用を起こすことがあるため、服用中のすべての薬剤を担当医に報告すること。また、本剤で治療中に新たに他の薬剤を服用する場合には、事前に担当医に報告すること。
8.4 肝機能障害があらわれることがあるので、定期的に肝機能検査を行う等、観察を十分に行うこと。[11.1.1参照]
9. 特定の背景を有する患者に関する注意
9.3 肝機能障害患者
9.3.1 重度の肝機能障害(Child-Pugh分類:C)患者
重度(Child-Pugh分類:C)の肝機能障害患者を対象とした臨床試験は実施していない。[11.1.1参照]
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。本剤は投与後に長期間(12ヵ月以上)にわたって血中に残留する可能性があるため、妊娠した場合に胎児が本剤に曝露される可能性がある。動物実験(ラット)において、1000mg/kg/日(最大臨床用量におけるヒト曝露量の26倍)の経口投与時に、胎児体重の低値、分娩遅延、死産数の増加及び出生児の生存率低下が報告されている。また、動物実験(ラット)で胎盤通過性が認められている。[8.5参照]
9.6 授乳婦
授乳を避けさせること。一般に、乳児へのHIV感染を防ぐため、あらゆる状況下においてHIVに感染した女性は授乳をすべきでない。また、本剤の最後の投与から長期間(12ヵ月以上)にわたって本剤が乳汁中に認められる可能性がある。動物実験(ラット)において、妊娠6日から分娩20日にカボテグラビルを経口投与したとき、生後10日の出生児血漿中に薬物が認められたことから、ヒトにおいても乳汁に移行する可能性がある。[8.5参照]
9.7 小児等
小児等を対象とした臨床試験は実施していない。
10. 相互作用
相互作用序文
薬物代謝酵素用語
UGT1A1
薬物代謝酵素用語
OAT1
薬物代謝酵素用語
OAT3
10.1 併用禁忌
| リファンピシン リファジン [2.2、16.7.3参照] | 本剤の血漿中濃度が低下し、本剤の効果が減弱するおそれがある。 | これらの薬剤がUGT1A1を誘導することにより、本剤の代謝が促進される。 |
| カルバマゼピン テグレトール [2.2参照] | 本剤の血漿中濃度が低下し、本剤の効果が減弱するおそれがある。 | これらの薬剤がUGT1A1を誘導することにより、本剤の代謝が促進される。 |
| フェニトイン アレビアチン [2.2参照] | 本剤の血漿中濃度が低下し、本剤の効果が減弱するおそれがある。 | これらの薬剤がUGT1A1を誘導することにより、本剤の代謝が促進される。 |
| ホスフェニトイン ホストイン [2.2参照] | 本剤の血漿中濃度が低下し、本剤の効果が減弱するおそれがある。 | これらの薬剤がUGT1A1を誘導することにより、本剤の代謝が促進される。 |
| フェノバルビタール フェノバール [2.2参照] | 本剤の血漿中濃度が低下し、本剤の効果が減弱するおそれがある。 | これらの薬剤がUGT1A1を誘導することにより、本剤の代謝が促進される。 |
10.2 併用注意
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 肝機能障害(頻度不明)
11.1.2 薬剤性過敏症症候群(頻度不明)
重度又は発熱を伴う発疹、全身倦怠感、疲労、筋肉痛又は関節痛、水疱、口腔病変、結膜炎、顔面浮腫、肝炎、好酸球増加症又は血管性浮腫等があらわれた場合には投与を中止し、肝機能検査を行う等、患者の状態を十分に観察すること。
リルピビリン製剤併用時の経口剤及び注射剤における発現頻度
11.2 その他の副作用
| 10%以上 | 1〜10%未満 | 1%未満 | 頻度不明 | |
| 精神・神経系 | 頭痛、不安、異常な夢、不眠症、浮動性めまい | うつ病、傾眠 | 血管迷走神経性反応、自殺念慮、自殺企図 | |
| 消化器 | 悪心、下痢 | 嘔吐、腹痛、鼓腸 | ||
| 皮膚 | 発疹 | 血管性浮腫、蕁麻疹 | ||
| 筋骨格 | 筋肉痛 | |||
| 投与部位 | 注射部位反応(疼痛、結節、硬結) | 注射部位反応(不快感、腫脹、紅斑、そう痒感、内出血、熱感、血腫、知覚消失) | 注射部位反応(蜂巣炎、膿瘍、出血、変色) | |
| 全身症状 | 発熱、疲労、無力症、倦怠感 | |||
| 臨床検査 | 体重増加、トランスアミナーゼ上昇、リパーゼ増加 | 総ビリルビン上昇 |
14. 適用上の注意
14.1 薬剤調製時の注意
14.1.1 バイアル内の懸濁液が均一になるまで約10秒間激しく振とうする。小さな気泡が見えることがあるが問題はない。
14.1.2 シリンジに採取後、ただちに使用しない場合は、室温で保存し、2時間以内に使用すること。2時間を超えて放置した場合は、廃棄すること。
14.2 薬剤投与時の注意
14.2.1 注射部位は、臀部の外側上部とすること。筋肉内にのみ投与し、静脈内には投与しないこと。
14.2.2 本剤とリルピビリン注射剤は、同日に臀部の筋肉の異なる部位(左右異なる側又は2cm以上間隔をあける)に投与すること。
14.2.3 本剤を投与する場合、21〜23ゲージの注射針の使用が推奨される。なお、注射針の長さは、BMIを考慮し、臀部の筋肉に到達するものを用いること。
14.2.4 注射部位での薬物漏出を最小限に抑えるため、Z-track法を用いて投与する。皮膚を一方向へ約2.5cm強く引いて保持し、針を筋肉に達するまで深く刺入して投与し、抜針後、速やかに引いていた皮膚を戻すこと。なお、注射部位を押さえるが、もまないこと。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
健康成人14例にカボテグラビル400mgを単回筋肉内投与注)した時の薬物動態パラメータ及び血漿中濃度推移をそれぞれ表-1及び図-1に示す1)(外国人データ)。
| AUC(0-t)(μg・h/mL) | Cmax(μg/mL) | tmax(日) | t1/2(日) |
| 1921.1 (1389.3,2656.6) | 0.7 (0.5,0.9) | 69.0 (2.0-213.0)注1) | 38.3 (26.2,56.1)注2) |
図-1 健康成人にカボテグラビル400mgを単回筋肉内投与した時の血漿中薬物濃度推移(平均値+標準偏差、14例)
16.1.2 反復投与
母集団薬物動態モデルを用いた、HIV感染症患者を対象とした国際共同第III相試験(201584試験及び201585試験)におけるカボテグラビルとリルピビリン注射剤投与時のカボテグラビルの薬物動態パラメータ(推定値)を日本人及び外国人集団別に表-2に示す。[17.1.1、17.1.2参照]
初回投与及び4週間隔で投与したときの薬物動態パラメータ(推定値)については、国際共同第III相試験における実測値データに基づく推定値であり、8週間隔で投与したときの薬物動態パラメータ(推定値)については、カボテグラビル注射剤を4週間隔で反復筋肉内投与した時の実測値に基づく各被験者の事後推定値を踏まえて予測した結果である。
初回投与及び4週間隔で投与したときの薬物動態パラメータ(推定値)については、国際共同第III相試験における実測値データに基づく推定値であり、8週間隔で投与したときの薬物動態パラメータ(推定値)については、カボテグラビル注射剤を4週間隔で反復筋肉内投与した時の実測値に基づく各被験者の事後推定値を踏まえて予測した結果である。
| 投与時期/投与間隔 | 患者 | 例数 | AUC(0-τ)(μg・h/mL) | Cmax(μg/mL) | tmax注1)(日) | Cτ(μg/mL) | |
| 初回投与 | 日本人 | 8 | 2516 (2086,3034) | 9.5 (8.6,10.6) | 0注2) (0-24) | 2.3 (2,2.7) | |
| 外国人 | 732 | 1556 (1505,1609) | 8.0 (7.9,8.2) | 0注2) (0-72) | 1.4 (1.4,1.5) | ||
| 4週間隔 | 定常状態 | 日本人 | 8 | 3067 (2693,3493) | 5.5 (4.9,6.3) | 5 (5-6) | 3.3 (2.8,4) |
| 外国人 | 732 | 2429 (2382,2477) | 4.2 (4.1,4.3) | 5 (4-7) | 2.9 (2.8,3) | ||
| 8週間隔 | 日本人 | 8 | 4613 (4047,5258) | 5.4 (4.7,6.2) | 6 (6-7) | 1.7 (1.2,2.3) | |
| 外国人 | 732 | 3691 (3620,3763) | 3.8 (3.7,3.9) | 6 (4-9) | 1.7 (1.6,1.7) | ||
16.3 分布
16.3.1 血漿蛋白結合率
In vitroでのカボテグラビルのヒト血漿蛋白結合率は99%超であった2)。
16.3.2 分布容積
カボテグラビルの見かけの中心コンパートメント及び末梢コンパートメントの分布容積はそれぞれ5.27L及び2.43Lであった(母集団薬物動態解析による推定値)。
16.3.3 血球移行性
ヒトでの血液:血漿の比(平均値)は0.437〜0.571であった3)。
16.3.4 脳脊髄液への移行
カボテグラビルは脳脊髄液中に分布する。HIV感染症患者にカボテグラビル400mgを4週間隔で、カボテグラビル600mgを8週間隔で筋肉内投与した時、定常状態における投与1週間後のカボテグラビルの脳脊髄液中濃度と血漿中濃度との比(中央値)はいずれも0.003であった4)(外国人データ)。
16.3.5 組織内分布
カボテグラビルは男性及び女性の生殖器に分布する。健康成人にカボテグラビル400mgを単回筋肉内投与した時、子宮頸部及び膣組織:血漿比の中央値は0.16〜0.28、直腸組織:血漿比の中央値は0.08以下であった1)(外国人データ)。
16.4 代謝
16.5 排泄
健康成人に14C-カボテグラビル30mg(水溶液)注)を単回経口投与した時の総投与量の約59%が糞中に、約27%が尿中に回収された。糞中排泄物の大部分(総投与量の約47%)は未変化体であり、尿中には代謝物のみ検出された3)(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
重度の腎機能低下者(8例、クレアチニンクリアランス(Ccr):30mL/min未満)及び健康成人8例にカボテグラビル30mgを単回経口投与注)した時の血漿中カボテグラビルの薬物動態パラメータを表-3に示す6)(外国人データ)。なお、透析患者での本剤の薬物動態に及ぼす影響については検討していない。
| 被験者 | 例数 | Cmax(μg/mL) | AUC(0-inf)(μg・h/mL) | t1/2(h) |
| 重度の腎機能低下者 | 8 | 3.34 (2.67,4.17) | 142.72注1) (115.40,176.51) | 39.24注1) (33.93,45.39) |
| 健康成人 | 8 | 3.37 (2.96,3.83) | 140.48 (115.84,170.37) | 40.54 (36.92,44.52) |
16.6.2 肝機能障害患者
中等度の肝機能低下者(8例、Child-Pugh分類:B)及び健康成人8例にカボテグラビル30mgを単回経口投与注)した時の血漿中カボテグラビルの薬物動態パラメータを表-4に示す7)(外国人データ)。なお、重度の肝機能低下者での本剤の薬物動態に及ぼす影響については検討していない。
| 被験者 | 例数 | Cmax(μg/mL) | AUC(0-inf)(μg・h/mL) | t1/2(h) |
| 中等度の肝機能低下者 | 8 | 2.70 (1.94,3.76) | 101.73 (75.22,137.58) | 30.85 (23.72,40.13) |
| 健康成人 | 8 | 3.55 (2.90,4.33) | 127.08 (94.74,170.47) | 37.25 (33.41,41.53) |
16.7 薬物相互作用
16.7.1 In vitro試験
16.7.2 カボテグラビルが併用薬の薬物動態に及ぼす影響
カボテグラビルが併用薬の薬物動態に及ぼす影響を表-5に示す(外国人データ)。
| 併用薬及び用量 | カボテグラビルの用量注1) | 例数 | カボテグラビル併用時/非併用時の併用薬の薬物動態パラメータの幾何平均の比 (90%信頼区間) | ||
| Cmax | AUC | Cτ又はC24 | |||
| エチニルエストラジオール 0.03mg 1日1回11) | 30mg注2) | 19 | 0.92 (0.83,1.03) | 1.02 (0.97,1.08) | 1.00 (0.92,1.10) |
| レボノルゲストレル 0.15mg 1日1回11) | 30mg注2) | 19 | 1.05 (0.96,1.15) | 1.12 (1.07,1.18) | 1.07 (1.01,1.15) |
| ミダゾラム 3mg 単回12) | 30mg注2) | 12 | 1.09 (0.94,1.26) | 1.08 (0.96,1.22) | − |
| リルピビリン 25mg 1日1回13) | 30mg注2) | 11 | 0.96 (0.85,1.09) | 0.99 (0.89,1.09) | 0.92 (0.79,1.07) |
16.7.3 併用薬がカボテグラビルの薬物動態に及ぼす影響
併用薬がカボテグラビルの薬物動態に及ぼす影響を表-6に示す(外国人データ)。[10.1、10.2参照]
| 併用薬及び用量 | カボテグラビルの用量注1) | 例数 | 他剤併用時/非併用時のカボテグラビルの薬物動態パラメータの幾何平均の比 (90%信頼区間) | ||
| Cmax | AUC | Cτ又はC24 | |||
| エトラビリン 200mg 1日2回14) | 30mg注2) | 12 | 1.04 (0.99,1.09) | 1.01 (0.96,1.06) | 1.00 (0.94,1.06) |
| リファブチン 300mg 1日1回15) | 30mg注2) | 12 | 0.83 (0.76,0.90) | 0.79 (0.74,0.83) | 0.74 (0.70,0.78) |
| リファンピシン 600mg 1日1回16) | 30mg注3) | 15 | 0.94 (0.87,1.02) | 0.41 (0.36,0.46) | 0.50 (0.44,0.57) |
| リルピビリン 25mg 1日1回13) | 30mg注2) | 11 | 1.05 (0.96,1.15) | 1.12 (1.05,1.19) | 1.14 (1.04,1.24) |
注)本剤の承認された用法及び用量は、「<1ヵ月間隔投与>リルピビリンとの併用において、通常、成人にはカボテグラビルとして600mgを臀部筋肉内に投与する。以降は、400mgを1ヵ月に1回、臀部筋肉内に投与する。<2ヵ月間隔投与>リルピビリンとの併用において、通常、成人にはカボテグラビルとして600mgを臀部筋肉内に投与する。本剤初回投与1ヵ月後に600mgを臀部筋肉内に投与し、以降は600mgを2ヵ月に1回、臀部筋肉内に投与する。」である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国際共同第III相試験(FLAIR:201584試験)
抗レトロウイルス療法による治療経験のない成人HIV-1感染症患者を対象にインテグラーゼ阻害剤(INSTI)を含む1日1回1錠のレジメンからカボテグラビルとリルピビリンの併用療法に切り替えた後のウイルス学的抑制の維持の評価を目的としたランダム化非盲検比較試験に629例が組み入れられた。組み入れられた被験者にドルテグラビル・アバカビル・ラミブジン配合錠[HLA-B*5701陽性被験者では、ドルテグラビルと核酸系逆転写酵素阻害剤(NRTI)2剤]を1日1回、20週間経口投与した。HIV-1 RNA量が50copies/mL未満であった被験者566例(日本人患者20例を含む)のうち、カボテグラビルとリルピビリンの併用投与群(CAB+RPV群)に283例、ドルテグラビル・アバカビル・ラミブジン配合錠[HLA-B*5701陽性被験者では、ドルテグラビルと核酸系逆転写酵素阻害剤(NRTI)2剤]を継続する群(継続投与群)に283例が割り付けられた。CAB+RPV群に割り付けられた被験者には、カボテグラビル経口剤30mgとリルピビリン経口剤25mgを1日1回、少なくとも4週間併用経口投与した後、カボテグラビル注射剤(1ヵ月目600mg、2ヵ月目以降400mg)とリルピビリン注射剤(1ヵ月目900mg、2ヵ月目以降600mg)を1ヵ月間隔で44週間臀部筋肉内に併用投与した17)。両群の患者背景及び疾患特性に偏りはみられずCAB+RPV群の年齢中央値は34歳(範囲19-68歳)、女性22%、人種は白人76%、黒人又はアフリカ系アメリカ人17%、アジア人4%、その他が3%であった。ベースラインのCD4陽性リンパ球数350cells/mm3未満は7%であった。
主要評価項目である投与48週時のHIV-1 RNA量が50copies/mL以上であった被験者の割合は、継続投与群の2.5%に対して、CAB+RPV群で2.1%であり、調整した群間差の95%信頼区間の上限値(2.1%)は、非劣性マージン(6%)より小さく、継続投与群に対するCAB+RPV群の非劣性が示された。48週時までにウイルス学的失敗の基準(HIV-1 RNA量が200copies/mL未満に抑制された後、2回の連続するHIV-1 RNA量の測定結果が200copies/mL以上)を満たした被験者はCAB+RPV群で1.4%(4/283例)、継続投与群で1.1%(3/283例)であった。48週時のベースライン特性別のHIV-1 RNA量が50copies/mL以上であった被験者の割合は、CAB+RPV群及び継続投与群で同程度であった。日本人集団における主要評価項目である投与48週時のHIV-1 RNA量が50copies/mL以上であった被験者は、CAB+RPV群(8例)及び継続投与群(12例)両群ともに0例であった。[5.1、16.1.2参照]
副作用発現頻度は、CAB+RPV群で83%(236/283例)であった。主な副作用は、注射部位疼痛78%(221/283例)、注射部位結節15%(43/283例)、注射部位硬結13%(37/283例)、注射部位腫脹8%(22/283例)、注射部位そう痒感6%(16/283例)、頭痛5%(14/283例)、発熱5%(13/283例)、注射部位紅斑4%(12/283例)、注射部位熱感3%(8/283例)及び体温上昇3%(8/283例)であった。日本人集団において2例以上にみられた副作用は、注射部位疼痛88%(7/8例)、倦怠感38%(3/8例)であった。
なお、本試験における試験成績の要約を表-1に、ベースラインの特性別の48週時のHIV-1 RNA量が50copies/mL以上の被験者の割合を表-2に示した。
主要評価項目である投与48週時のHIV-1 RNA量が50copies/mL以上であった被験者の割合は、継続投与群の2.5%に対して、CAB+RPV群で2.1%であり、調整した群間差の95%信頼区間の上限値(2.1%)は、非劣性マージン(6%)より小さく、継続投与群に対するCAB+RPV群の非劣性が示された。48週時までにウイルス学的失敗の基準(HIV-1 RNA量が200copies/mL未満に抑制された後、2回の連続するHIV-1 RNA量の測定結果が200copies/mL以上)を満たした被験者はCAB+RPV群で1.4%(4/283例)、継続投与群で1.1%(3/283例)であった。48週時のベースライン特性別のHIV-1 RNA量が50copies/mL以上であった被験者の割合は、CAB+RPV群及び継続投与群で同程度であった。日本人集団における主要評価項目である投与48週時のHIV-1 RNA量が50copies/mL以上であった被験者は、CAB+RPV群(8例)及び継続投与群(12例)両群ともに0例であった。[5.1、16.1.2参照]
副作用発現頻度は、CAB+RPV群で83%(236/283例)であった。主な副作用は、注射部位疼痛78%(221/283例)、注射部位結節15%(43/283例)、注射部位硬結13%(37/283例)、注射部位腫脹8%(22/283例)、注射部位そう痒感6%(16/283例)、頭痛5%(14/283例)、発熱5%(13/283例)、注射部位紅斑4%(12/283例)、注射部位熱感3%(8/283例)及び体温上昇3%(8/283例)であった。日本人集団において2例以上にみられた副作用は、注射部位疼痛88%(7/8例)、倦怠感38%(3/8例)であった。
なお、本試験における試験成績の要約を表-1に、ベースラインの特性別の48週時のHIV-1 RNA量が50copies/mL以上の被験者の割合を表-2に示した。
| CAB+RPV群 283例 | 継続投与群 283例 | |
| HIV-1 RNA量が50copies/mL以上注1) | 6例(2.1%) | 7例(2.5%) |
| 両群間の差(95%信頼区間)注2) | −0.4%(−2.8%,2.1%) | |
| ウイルス学的失敗注3) | 4例(1.4%)注4) | 3例(1.1%) |
| CAB+RPV群 283例 | 継続投与群 283例 | |
| ベースラインCD4陽性リンパ球数(cells/mm2) | ||
| <350 | 0/19 | 1/27(3.7%) |
| 350-<500 | 3/64(4.7%) | 0/60 |
| ≧500 | 3/200(1.5%) | 6/196(3.1%) |
| 性別 | ||
| 男性 | 3/220(1.4%) | 6/219(2.7%) |
| 女性 | 3/63(4.8%) | 1/64(1.6%) |
| 人種 | ||
| 白人 | 6/216(2.8%) | 5/201(2.5%) |
| 黒人/アフリカ系アメリカ人 | 0/47 | 2/56(3.6%) |
| アジア人 | 0/12 | 0/15 |
| その他 | 0/8 | 0/9 |
| BMI(kg/m2) | ||
| <30 | 3/243(1.2%) | 7/246(2.8%) |
| ≧30 | 3/40(7.5%) | 0/37 |
| 年齢(歳) | ||
| <50 | 5/250(2.0%) | 6/254(2.4%) |
| ≧50 | 1/33(3.0%) | 1/29(3.4%) |
17.1.2 海外第III相試験(ATLAS:201585試験)
抗レトロウイルス療法により、少なくとも6ヵ月間ウイルス学的に抑制されている成人HIV-1感染症患者616例を対象としたランダム化非盲検試験において、カボテグラビルとリルピビリンの併用投与群(CAB+RPV群)に308例、現行のレジメンを継続する群(継続投与群)に308例が割り付けられた。CAB+RPV群に割り付けられた被験者には、カボテグラビル経口剤30mgとリルピビリン経口剤25mgを1日1回、少なくとも4週間併用経口投与した後、カボテグラビル注射剤(1ヵ月目600mg、2ヵ月目以降400mg)とリルピビリン注射剤(1ヵ月目900mg、2ヵ月目以降600mg)を1ヵ月間隔で44週間臀部筋肉内に併用投与した18)。両群の患者背景及び疾患特性に偏りはみられずCAB+RPV群の年齢中央値は40歳(範囲21-74歳)、女性32%、人種は白人69%、黒人又はアフリカ系アメリカ人20%、アジア人7%、その他が3%であった。ベースラインのCD4陽性リンパ球数350cells/mm3未満は7%であった。
主要評価項目である投与48週時のHIV-1 RNA量が50copies/mL以上であった被験者の割合は、継続投与群の1.0%に対して、CAB+RPV群で1.6%であり、調整した群間差の95%信頼区間の上限値(2.5%)は、非劣性マージン(6%)より小さく、継続投与群に対するCAB+RPV群の非劣性が示された。48週時までにウイルス学的失敗の基準(HIV-1 RNA量が200copies/mL未満に抑制された後、2回の連続するHIV-1 RNA量の測定結果が200copies/mL以上)を満たした被験者はCAB+RPV群で1.0%(3/308例)、継続投与群で1.3%(4/308例)であった。48週時のベースライン特性別のHIV-1 RNA量が50copies/mL以上であった被験者の割合は、CAB+RPV群及び継続投与群で同程度であった。[5.1、16.1.2参照]
副作用発現頻度は、CAB+RPV群で83%(255/308例)であった。主な副作用は、注射部位疼痛74%(227/308例)、注射部位結節12%(36/308例)、注射部位硬結9%(29/308例)、注射部位腫脹7%(22/308例)、注射部位紅斑4%(12/308例)、疲労4%(11/308例)、発熱4%(11/308例)、注射部位内出血3%(10/308例)、悪心4%(11/308例)、頭痛4%(11/308例)及び不眠症3%(8/308例)であった。
なお、本試験における試験成績の要約を表-3に、ベースラインの特性別の48週時のHIV-1 RNA量が50copies/mL以上の被験者の割合を表-4に示した。
主要評価項目である投与48週時のHIV-1 RNA量が50copies/mL以上であった被験者の割合は、継続投与群の1.0%に対して、CAB+RPV群で1.6%であり、調整した群間差の95%信頼区間の上限値(2.5%)は、非劣性マージン(6%)より小さく、継続投与群に対するCAB+RPV群の非劣性が示された。48週時までにウイルス学的失敗の基準(HIV-1 RNA量が200copies/mL未満に抑制された後、2回の連続するHIV-1 RNA量の測定結果が200copies/mL以上)を満たした被験者はCAB+RPV群で1.0%(3/308例)、継続投与群で1.3%(4/308例)であった。48週時のベースライン特性別のHIV-1 RNA量が50copies/mL以上であった被験者の割合は、CAB+RPV群及び継続投与群で同程度であった。[5.1、16.1.2参照]
副作用発現頻度は、CAB+RPV群で83%(255/308例)であった。主な副作用は、注射部位疼痛74%(227/308例)、注射部位結節12%(36/308例)、注射部位硬結9%(29/308例)、注射部位腫脹7%(22/308例)、注射部位紅斑4%(12/308例)、疲労4%(11/308例)、発熱4%(11/308例)、注射部位内出血3%(10/308例)、悪心4%(11/308例)、頭痛4%(11/308例)及び不眠症3%(8/308例)であった。
なお、本試験における試験成績の要約を表-3に、ベースラインの特性別の48週時のHIV-1 RNA量が50copies/mL以上の被験者の割合を表-4に示した。
| CAB+RPV群 308例 | 継続投与群 308例 | |
| HIV-1 RNA量が50copies/mL以上注1) | 5例(1.6%) | 3例(1.0%) |
| 両群間の差(95%信頼区間)注2) | 0.6%(−1.2%,2.5%) | |
| ウイルス学的失敗注3) | 3例(1.0%)注4) | 4例(1.3%) |
| CAB+RPV群 308例 | 継続投与群 308例 | |
| ベースラインCD4陽性リンパ球数(cells/mm3) | ||
| <350 | 0/23 | 1/27(3.7%) |
| 350-<500 | 2/56(3.6%) | 0/57 |
| ≧500 | 3/229(1.3%) | 2/224(0.9%) |
| 性別 | ||
| 男性 | 3/209(1.4%) | 3/204(1.5%) |
| 女性 | 2/99(2.0%) | 0/104 |
| 人種 | ||
| 白人 | 3/214(1.4%) | 2/207(1.0%) |
| 黒人/アフリカ系アメリカ人 | 2/62(3.2%) | 1/77(1.3%) |
| アジア人 | 0/22 | 0/13 |
| その他 | 0/10 | 0/11 |
| BMI(kg/m2) | ||
| <30 | 3/248(1.2%) | 1/242(0.4%) |
| ≧30 | 2/60(3.3%) | 2/66(3.0%) |
| 年齢(歳) | ||
| <50 | 4/242(1.7%) | 2/212(0.9%) |
| ≧50 | 1/66(1.5%) | 1/96(1.0%) |
| ランダム化時の継続投与 | ||
| PI | 1/51(2.0%) | 0/54 |
| INSTI | 0/102 | 2/99(2.0%) |
| NNRTI | 4/155(2.6%) | 1/155(0.6%) |
17.1.3 海外第III相試験(ATLAS-2M:207966試験)
抗レトロウイルス療法により、ウイルス学的に抑制されている成人HIV-1感染症患者1045例を対象としたランダム化非盲検試験において、カボテグラビルとリルピビリンを1ヵ月間隔で併用投与する群(1ヵ月間隔投与群)に523例、2ヵ月間隔で併用投与する群(2ヵ月間隔投与群)に522例が割り付けられた。
割付け前にカボテグラビルとリルピビリンの併用療法以外の治療を受けていた被験者には、カボテグラビル経口剤30mgとリルピビリン経口剤25mgを1日1回、少なくとも4週間併用経口投与した。1ヵ月間隔投与群では、カボテグラビル注射剤(1ヵ月目600mg、2ヵ月目以降1ヵ月間隔で400mg)とリルピビリン注射剤(1ヵ月目900mg、2ヵ月目以降1ヵ月間隔で600mg)を44週間臀部筋肉内に併用投与した。2ヵ月間隔投与群では、カボテグラビル注射剤(1、2ヵ月目及び以降2ヵ月間隔で600mg)とリルピビリン注射剤(1、2ヵ月目及び以降2ヵ月間隔で900mg)を44週間臀部筋肉内に併用投与した19)。1ヵ月間隔投与群及び2ヵ月間隔投与群の患者背景及び疾患特性に偏りはみられず、年齢の中央値はいずれも42.0歳、性別は両群ともに男性が70%以上で、人種も70%以上が白人であり、CD4陽性リンパ球数350cells/mm3未満は、それぞれ5%及び7%であった。
主要評価項目である投与48週時のHIV-1 RNA量が50copies/mL以上であった被験者の割合は、1ヵ月間隔投与群の1.0%に対して、2ヵ月間隔投与群で1.7%であり、調整した群間差の95%信頼区間の上限値(2.2%)は、非劣性マージン(4%)より小さく、1ヵ月間隔投与群に対する2ヵ月間隔投与群の非劣性が示された。48週時までにウイルス学的失敗の基準(HIV-1 RNA量が200copies/mL未満に抑制された後、2回の連続するHIV-1 RNA量の測定結果が200copies/mL以上)を満たした被験者は1ヵ月間隔投与群で0.4%(2/523例)、2ヵ月間隔投与群で1.5%(8/522例)であった。48週時のベースライン特性別のHIV-1 RNA量が50copies/mL以上であった被験者の割合は、両群で同程度であった。[5.1、7.3参照]
副作用発現頻度は、1ヵ月間隔投与群で76%(399/523例)、2ヵ月間隔投与群で77%(400/522例)であった。1ヵ月間隔投与群の主な副作用は、注射部位疼痛68%(358/523例)、注射部位結節17%(87/523例)、注射部位硬結7%(37/523例)、注射部位不快感8%(40/523例)、注射部位腫脹5%(26/523例)、発熱5%(25/523例)、注射部位そう痒感5%(24/523例)、疲労4%(19/523例)、注射部位紅斑3%(15/523例)及び注射部位血腫3%(14/523例)であり、2ヵ月間隔投与群の主な副作用は、注射部位疼痛70%(364/522例)、注射部位結節10%(54/522例)、注射部位硬結8%(40/522例)、注射部位不快感7%(34/522例)、注射部位腫脹6%(32/522例)、注射部位そう痒感5%(26/522例)及び発熱4%(19/522例)であった。
なお、本試験における試験成績の要約を表-5に、ベースラインの特性別の48週時のHIV-1 RNA量が50copies/mL以上の被験者の割合を表-6に示した。
割付け前にカボテグラビルとリルピビリンの併用療法以外の治療を受けていた被験者には、カボテグラビル経口剤30mgとリルピビリン経口剤25mgを1日1回、少なくとも4週間併用経口投与した。1ヵ月間隔投与群では、カボテグラビル注射剤(1ヵ月目600mg、2ヵ月目以降1ヵ月間隔で400mg)とリルピビリン注射剤(1ヵ月目900mg、2ヵ月目以降1ヵ月間隔で600mg)を44週間臀部筋肉内に併用投与した。2ヵ月間隔投与群では、カボテグラビル注射剤(1、2ヵ月目及び以降2ヵ月間隔で600mg)とリルピビリン注射剤(1、2ヵ月目及び以降2ヵ月間隔で900mg)を44週間臀部筋肉内に併用投与した19)。1ヵ月間隔投与群及び2ヵ月間隔投与群の患者背景及び疾患特性に偏りはみられず、年齢の中央値はいずれも42.0歳、性別は両群ともに男性が70%以上で、人種も70%以上が白人であり、CD4陽性リンパ球数350cells/mm3未満は、それぞれ5%及び7%であった。
主要評価項目である投与48週時のHIV-1 RNA量が50copies/mL以上であった被験者の割合は、1ヵ月間隔投与群の1.0%に対して、2ヵ月間隔投与群で1.7%であり、調整した群間差の95%信頼区間の上限値(2.2%)は、非劣性マージン(4%)より小さく、1ヵ月間隔投与群に対する2ヵ月間隔投与群の非劣性が示された。48週時までにウイルス学的失敗の基準(HIV-1 RNA量が200copies/mL未満に抑制された後、2回の連続するHIV-1 RNA量の測定結果が200copies/mL以上)を満たした被験者は1ヵ月間隔投与群で0.4%(2/523例)、2ヵ月間隔投与群で1.5%(8/522例)であった。48週時のベースライン特性別のHIV-1 RNA量が50copies/mL以上であった被験者の割合は、両群で同程度であった。[5.1、7.3参照]
副作用発現頻度は、1ヵ月間隔投与群で76%(399/523例)、2ヵ月間隔投与群で77%(400/522例)であった。1ヵ月間隔投与群の主な副作用は、注射部位疼痛68%(358/523例)、注射部位結節17%(87/523例)、注射部位硬結7%(37/523例)、注射部位不快感8%(40/523例)、注射部位腫脹5%(26/523例)、発熱5%(25/523例)、注射部位そう痒感5%(24/523例)、疲労4%(19/523例)、注射部位紅斑3%(15/523例)及び注射部位血腫3%(14/523例)であり、2ヵ月間隔投与群の主な副作用は、注射部位疼痛70%(364/522例)、注射部位結節10%(54/522例)、注射部位硬結8%(40/522例)、注射部位不快感7%(34/522例)、注射部位腫脹6%(32/522例)、注射部位そう痒感5%(26/522例)及び発熱4%(19/522例)であった。
なお、本試験における試験成績の要約を表-5に、ベースラインの特性別の48週時のHIV-1 RNA量が50copies/mL以上の被験者の割合を表-6に示した。
| 1ヵ月間隔投与群 523例 | 2ヵ月間隔投与群 522例 | |
| HIV-1 RNA量が50copies/mL以上注1) | 5例(1.0%) | 9例(1.7%) |
| 両群間の差(95%信頼区間)注2) | 0.8%(−0.6%,2.2%) | |
| ウイルス学的失敗注3) | 2例(0.4%)注4) | 8例(1.5%)注4) |
| 1ヵ月間隔投与群 523例 | 2ヵ月間隔投与群 522例 | |
| ベースラインCD4陽性リンパ球数(cells/mm3) | ||
| <350 | 1/27(3.7%) | 1/35(2.9%) |
| 350-<500 | 0/89 | 1/96(1.0%) |
| ≧500 | 4/407(1.0%) | 7/391(1.8%) |
| 性別 | ||
| 男性 | 5/380(1.3%) | 4/385(1.0%) |
| 女性 | 0/143 | 5/137(3.6%) |
| 人種 | ||
| 白人 | 5/393(1.3%) | 5/370(1.4%) |
| 非白人 | 0/130 | 4/152(2.6%) |
| 黒人/アフリカ系アメリカ人 | 0/90 | 4/101(4.0%) |
| 非黒人/アフリカ系アメリカ人 | 5/433(1.2%) | 5/421(1.2%) |
| BMI(kg/m2) | ||
| <30 | 3/425(0.7%) | 3/409(0.7%) |
| ≧30 | 2/98(2.0%) | 6/113(5.3%) |
| 年齢(歳) | ||
| <35 | 1/145(0.7%) | 4/137(2.9%) |
| 35-<50 | 2/239(0.8%) | 3/242(1.2%) |
| ≧50 | 2/139(1.4%) | 2/143(1.4%) |
| CAB+RPV投与歴(週) | ||
| 0 | 5/327(1.5%) | 5/327(1.5%) |
| 1-24 | 0/68 | 3/69(4.3%) |
| >24 | 0/128 | 1/126(0.8%) |
18. 薬効薬理
18.1 作用機序
カボテグラビルはレトロウイルスの複製に必要な酵素であるHIVインテグラーゼの活性部位に結合してその活性を阻害し、ウイルスDNAの宿主DNAへの組込みを抑制する。
18.2 抗ウイルス作用
野生型HIV-1 Ba-L株を感染させた末梢血単核球を用いた時のカボテグラビルのウイルス複製に対する50%阻害濃度(IC50)は0.22nMであった。24種類のHIV-1臨床分離株[グループM(サブタイプA、B、C、D、E、F及びG;それぞれ3種類ずつ)及びグループO;3種類]を感染させた細胞を用いた時のカボテグラビルのIC50は0.02〜1.06nMであり、3種類のHIV-2臨床分離株に対するIC50は0.10〜0.14nMであった。
HIV-1 IIIB株を感染させたMT-4細胞において、カボテグラビルをリルピビリン、ラミブジン、テノホビル又はエムトリシタビンと併用した時の抗ウイルス活性には、いずれも相加又は相乗効果が認められた。
HIV-1 IIIB株を感染させたMT-4細胞において、カボテグラビルをリルピビリン、ラミブジン、テノホビル又はエムトリシタビンと併用した時の抗ウイルス活性には、いずれも相加又は相乗効果が認められた。
18.3 薬剤耐性
18.3.1 非臨床試験成績
HIV-1 IIIB株(T124A多型を有する)をカボテグラビル存在下で112日間継代培養した試験で新たに認められたインテグラーゼ領域のアミノ酸変異はQ146L、S153Y及びI162Mであり、感受性変化度[Fold Change(FC):各変異を有する株に対するIC50/野生型HIV-1 NL432株に対するIC50]はそれぞれ1.3〜4.6、2.8〜8.4及び2.8であった。野生型HIV-1 NL432株をカボテグラビルの存在下で56日間継代培養した試験ではインテグラーゼ領域にアミノ酸変異は認められなかった。
18.3.2 臨床試験成績
201584(FLAIR)試験のCAB+RPV群において、耐性データの得られたウイルス学的失敗例3例中2例では、治療中にINSTI耐性関連Q148R変異を生じており、1例ではカボテグラビルに対する感受性低下を示すG140R変異が生じた。また、3例すべての被験者で1種類のリルピビリン耐性関連変異(K101E、E138E/A/K/T又はE138K)を生じており、3例中2例でリルピビリンに対する感受性の低下を示した。
201585(ATLAS)試験のウイルス学的失敗例3例中1例ではウイルス学的失敗の疑い時にINSTI耐性関連N155H変異が検出された。また、3例すべての被験者で治療中にリルピビリン耐性関連変異(E138A、E138E/K又はE138K)を生じており、リルピビリンに対する感受性の低下を示し、3例中1例はカボテグラビルに対する感受性の低下を示した。
カボテグラビルに対する耐性関連変異は、G140R(1例)、Q148R(2例)及びN155H(1例)であった。
カボテグラビルに対する耐性関連変異は、G140R(1例)、Q148R(2例)及びN155H(1例)であった。
207966(ATLAS-2M)試験において、1ヵ月間隔投与群のウイルス学的失敗例(2例)では、いずれの被験者もベースライン時にリルピビリン又はINSTI耐性関連変異を有していなかった。1例で非核酸系逆転写酵素阻害剤(NNRTI)関連変異(G190Q)とNNRTI多型(V189I)が同時に検出された。ウイルス学的失敗の疑い時に1例で治療中にリルピビリン耐性関連変異(K101E+M230L)が検出され、別の被験者ではNNRTI関連変異(G190Q+V189I)にV179V/Iが追加されていた。いずれの被験者においてもリルピビリンに対する感受性の低下を示した。また、いずれの被験者もウイルス学的失敗の疑い時にINSTI耐性関連変異(Q148R+E138E/K又はN155N/H)を有しており、1例ではカボテグラビルに対する感受性の低下を示した。いずれもINSTI関連変異であるL74Iは有しておらず、これらの被験者におけるカボテグラビルの感受性変化度は1.8〜4.6であった。2ヵ月間隔投与群のウイルス学的失敗例(8例)において、ベースライン時に5例がリルピビリン耐性関連変異(Y181Y/C+H221H/Y、Y188Y/F/H/L、Y188L、E138A又はE138E/A)を有し、1例がカボテグラビル耐性関連変異(G140G/R)を有していた(リルピビリン耐性関連変異Y181Y/F/H/Lを有していた症例と同一)。ウイルス学的失敗の疑い時に6例がリルピビリン耐性関連変異を有しており、うち2例でK101E、1例でE138E/Kがベースライン時から追加されていた。リルピビリンの感受性変化度は7例の被験者で生物学的カットオフ値を上回っていた(範囲:2.4〜15)。リルピビリン耐性関連変異を有していた6例中5例がINSTI耐性関連変異[N155H(2例)、Q148R(1例)及びQ148Q/R+N155N/H(2例)]を有していた。INSTI耐性関連変異であるL74Iが7例中4例の被験者でみられた。1例の被験者は、インテグラーゼ遺伝子型及び表現型アッセイの結果が得られず、他の1例ではカボテグラビル表現型の結果が得られなかった。これらの被験者におけるカボテグラビルの感受性変化度の範囲は0.6〜9.1であった。
18.4 交差耐性
INSTIに対する耐性関連変異(G118R、Q148K、Q148R、T66K/L74M、E92Q/N155H、E138A/Q148R、E138K/Q148K/R、G140C/Q148R、G140S/Q148H/K/R、Y143H/N155H及びQ148R/N155H)を導入したHIV-1 NL432株において、カボテグラビルに対する感受性の低下(野生型NL432株に対するIC50と比較することにより算出した感受性変化度が5以上)が認められた。そのうち、Q148K又はQ148Rを含む複数の変異を導入した場合に顕著な感受性の低下が認められ、N155H/Q148R及びE138K/Q148Kでの感受性変化度はそれぞれ61及び81であった。
カボテグラビルはNNRTI耐性関連変異(K103N及びY188L)及びNRTI耐性関連変異(M184V、D67N/K70R/T215Y及びV75I/F77L/F116Y/Q151M)を有する変異株に対して抗ウイルス活性を示した。
カボテグラビルはNNRTI耐性関連変異(K103N及びY188L)及びNRTI耐性関連変異(M184V、D67N/K70R/T215Y及びV75I/F77L/F116Y/Q151M)を有する変異株に対して抗ウイルス活性を示した。
19. 有効成分に関する理化学的知見
19.1. カボテグラビル
| 一般的名称 | カボテグラビル |
|---|---|
| 一般的名称(欧名) | Cabotegravir |
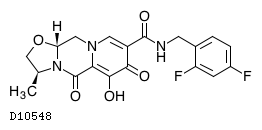
| 化学名 | (3S,11aR)-N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydrooxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide |
| 分子式 | C19H17F2N3O5 |
| 分子量 | 405.35 |
| 融点 | 約250℃ |
| 物理化学的性状 | 白色の固体 |
| KEGG DRUG |
|
20. 取扱い上の注意
凍結を避けて保存すること。
21. 承認条件
21.1 医薬品リスク管理計画を策定の上、適切に実施すること。
21.2 本剤の使用に当たっては、患者に対して本剤に関して更なる有効性・安全性のデータを引き続き収集中であること等を十分に説明し、インフォームドコンセントを得るよう、医師に要請すること。
21.3 現在実施中又は計画中の臨床試験については、終了後速やかに試験成績及び解析結果を提出すること。
21.4 再審査期間が終了するまでの間、原則として国内の全投与症例を対象とした製造販売後調査を実施し、本剤の使用実態に関する情報(患者背景、有効性・安全性(他剤併用時の有効性・安全性を含む)及び薬物相互作用のデータ等)を収集して定期的に報告するとともに、調査の結果を再審査申請時に提出すること。
22. 包装
<ボカブリア水懸筋注400mg>
2mL[1バイアル]
<ボカブリア水懸筋注600mg>
3mL[1バイアル]
23. 主要文献
- 社内資料:海外臨床試験(LAI114433、2022年5月31日承認、CTD2.7.2.2.3.1.3、2.7.2.2.3.1.4)
- 社内資料:分布に関する試験(2015N235936、2022年5月31日承認、CTD2.6.4.4.1.1)
- 社内資料:海外臨床試験(LAI117008、2022年5月31日承認、CTD2.7.2.1.2、2.7.2.2.6.1.2)
- 社内資料:海外臨床試験(200056、2022年5月31日承認、CTD2.7.2.2.1.2.5)
- 社内資料:代謝に関する試験(2012N145430、2022年5月31日承認、CTD2.6.4.5.1.1.5)
- 社内資料:海外臨床試験(201480、2022年5月31日承認、CTD2.7.2.2.6.5.2)
- 社内資料:海外臨床試験(201479、2022年5月31日承認、CTD2.7.2.2.6.5.1)
- 社内資料:分布に関する試験(2012N146040、2022年5月31日承認、CTD2.6.4.4.1.2)
- 社内資料:分布に関する試験(2012N155942、2022年5月31日承認、CTD2.6.4.4.1.4)
- 社内資料:分布に関する試験(2013N174474、2022年5月31日承認、CTD2.6.4.4.1.7)
- 社内資料:海外臨床試験(LAI117011、2022年5月31日承認、CTD2.7.2.2.6.6.4)
- 社内資料:海外臨床試験(LAI116815、2022年5月31日承認、CTD2.7.2.2.6.6.5)
- 社内資料:海外臨床試験(LAI116181、2022年5月31日承認、CTD2.7.2.2.6.6.1)
- 社内資料:海外臨床試験(ITZ111839、2022年5月31日承認、CTD2.7.2.2.6.6.3)
- 社内資料:海外臨床試験(205712、2022年5月31日承認、CTD2.7.2.2.6.6.2)
- 社内資料:海外臨床試験(LAI117010、2022年5月31日承認、CTD2.7.2.2.6.6.3)
- Orkin C,et al., N Engl J Med., 382 (12), 1124-1135, (2020) »PubMed
- Swindells S,et al., N Engl J Med., 382 (12), 1112-1123, (2020) »PubMed
- Overton ET,et al., Lancet., 396, 1994-2005, (2020) »PubMed
24. 文献請求先及び問い合わせ先
文献請求先
グラクソ・スミスクライン株式会社
ヴィーブヘルスケア・カスタマー・サービス
東京都港区赤坂1-8-1
電話:0120-066-525 (9:00〜17:45/土日祝日及び当社休業日を除く)
製品情報問い合わせ先
グラクソ・スミスクライン株式会社
ヴィーブヘルスケア・カスタマー・サービス
東京都港区赤坂1-8-1
電話:0120-066-525 (9:00〜17:45/土日祝日及び当社休業日を除く)
26. 製造販売業者等
26.1 製造販売元
ヴィーブヘルスケア株式会社
東京都港区赤坂1-8-1
26.2 販売元
グラクソ・スミスクライン株式会社
東京都港区赤坂1-8-1