医薬品情報
| 総称名 | エスシタロプラム |
|---|---|
| 一般名 | エスシタロプラムシュウ酸塩 |
| 欧文一般名 | Escitalopram Oxalate |
| 製剤名 | エスシタロプラムシュウ酸塩錠・エスシタロプラムシュウ酸塩口腔内崩壊錠 |
| 薬効分類名 | 選択的セロトニン再取り込み阻害剤(SSRI) |
| 薬効分類番号 | 1179 |
| ATCコード | N06AB10 |
| KEGG DRUG |
D02567
エスシタロプラムシュウ酸塩
|
| KEGG DGROUP |
DG01659
選択的セロトニン再取り込み阻害薬 (SSRI)
|
| JAPIC | 添付文書(PDF) |
添付文書情報2024年3月 改訂(第3版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| エスシタロプラム錠10mg「トーワ」 (後発品) | ESCITALOPRAM TABLETS 10mg"TOWA" | 東和薬品 | 1179054F1073 | 44.1円/錠 | 劇薬, 処方箋医薬品注) |
| エスシタロプラム錠20mg「トーワ」 (後発品) | ESCITALOPRAM TABLETS 20mg"TOWA" | 東和薬品 | 1179054F2070 | 65.6円/錠 | 劇薬, 処方箋医薬品注) |
| エスシタロプラムOD錠10mg「トーワ」 (後発品) | ESCITALOPRAM OD TABLETS 10mg"TOWA" | 東和薬品 | 1179054F3041 | 44.1円/錠 | 劇薬, 処方箋医薬品注) |
| エスシタロプラムOD錠20mg「トーワ」 (後発品) | ESCITALOPRAM OD TABLETS 20mg"TOWA" | 東和薬品 | 1179054F4048 | 65.6円/錠 | 劇薬, 処方箋医薬品注) |
2. 禁忌
次の患者には投与しないこと
4. 効能または効果
5. 効能または効果に関連する注意
<効能共通>
<うつ病・うつ状態>
5.2 本剤を12歳未満の大うつ病性障害患者に投与する際には適応を慎重に検討すること。[9.7.2参照]
<社会不安障害>
5.3 社会不安障害の診断は、DSM注1)等の適切な診断基準に基づき慎重に実施し、基準を満たす場合にのみ投与すること。
注1)DSM:American Psychiatric Association(米国精神医学会)のDiagnostic and Statistical Manual of Mental Disorders(精神疾患の診断・統計マニュアル)
6. 用法及び用量
通常、成人にはエスシタロプラムとして10mgを1日1回夕食後に経口投与する。なお、年齢・症状により適宜増減するが、増量は1週間以上の間隔をあけて行い、1日最高用量は20mgを超えないこととする。
7. 用法及び用量に関連する注意
7.1 本剤の投与量は必要最小限となるよう、患者ごとに慎重に観察しながら投与すること。
8. 重要な基本的注意
8.1 うつ症状を呈する患者は希死念慮があり、自殺企図のおそれがあるので、このような患者は投与開始早期ならびに投与量を変更する際には患者の状態及び病態の変化を注意深く観察すること。[5.1、8.2-8.4、9.1.3、9.1.4、15.1.1参照]
8.2 不安、焦燥、興奮、パニック発作、不眠、易刺激性、敵意、攻撃性、衝動性、アカシジア/精神運動不穏、軽躁、躁病等があらわれることが報告されている。また、因果関係は明らかではないが、これらの症状・行動を来した症例において、基礎疾患の悪化又は自殺念慮、自殺企図、他害行為が報告されている。患者の状態及び病態の変化を注意深く観察するとともに、これらの症状の増悪が観察された場合には、服薬量を増量せず、徐々に減量し、中止するなど適切な処置を行うこと。[5.1、8.1、8.3、8.4、9.1.3-9.1.6、15.1.1参照]
8.3 自殺目的での過量服用を防ぐため、自殺傾向が認められる患者に処方する場合には、1回分の処方日数を最小限にとどめること。[5.1、8.1、8.2、8.4、9.1.3、9.1.4、15.1.1参照]
8.4 家族等に自殺念慮や自殺企図、興奮、攻撃性、易刺激性等の行動の変化及び基礎疾患悪化があらわれるリスク等について十分説明を行い、医師と緊密に連絡を取り合うよう指導すること。[5.1、8.1-8.3、9.1.3-9.1.6、15.1.1参照]
8.5 眠気、めまい等があらわれることがあるので、本剤投与中の患者には、自動車の運転等危険を伴う機械を操作する際には十分注意させること。
8.6 投与中止(突然の中止)により、不安、焦燥、興奮、浮動性めまい、錯感覚、頭痛及び悪心等があらわれることが報告されている。投与を中止する場合には、突然の中止を避け、患者の状態を観察しながら徐々に減量すること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
(1)著明な徐脈等の不整脈又はその既往歴のある患者
(2)うっ血性心不全の患者
(3)低カリウム血症の患者
9.1.3 自殺念慮又は自殺企図の既往のある患者、自殺念慮のある患者
9.1.4 躁うつ病患者
9.1.5 脳の器質的障害又は統合失調症の素因のある患者
9.1.6 衝動性が高い併存障害を有する患者
9.1.7 てんかん等の痙攣性疾患又はこれらの既往歴のある患者
痙攣発作を起こすことがある。[11.1.1参照]
9.1.8 出血の危険性を高める薬剤を併用している患者、出血傾向又は出血性素因のある患者
出血傾向が増強するおそれがある。[10.2参照]
9.1.9 閉塞隅角緑内障の患者
眼圧上昇を起こし、症状が悪化するおそれがある。
9.2 腎機能障害患者
9.2.1 高度の腎機能障害のある患者
本剤のクリアランスが低下し、血中濃度が上昇するおそれがある。[16.6.1参照]
9.3 肝機能障害患者
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。
9.5.1 生殖発生毒性試験(ラット)において、臨床曝露量を超える高い曝露により胎児毒性(体重減少、骨化遅延)及び出生児の死亡率の増加が認められた。なお、動物実験(ラット)において、催奇形作用は認められていない。
9.5.2 本剤のラセミ体であるシタロプラムの生殖発生毒性試験(ラット)において、心血管系の異常を有する胎児数の増加が認められたが、再試験においては認められなかった。
9.5.3 妊娠末期に本剤あるいは他のSSRI、SNRIを投与された妊婦から出生した新生児において、入院期間の延長、呼吸補助、経管栄養を必要とする、離脱症状と同様の症状が出産直後にあらわれたとの報告がある。臨床所見としては、呼吸窮迫、チアノーゼ、無呼吸、発作、体温調節障害、哺乳障害、嘔吐、低血糖症、筋緊張低下、筋緊張亢進、反射亢進、振戦、ぴくつき、易刺激性、持続性の泣きが報告されている。
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。ヒト母乳中へ移行することが報告されている。
9.7 小児等
9.7.1 小児等を対象とした有効性及び安全性を指標とした臨床試験は実施していない。
9.8 高齢者
10. 相互作用
相互作用序文
本剤は主にCYP2C19で代謝され、CYP2D6及びCYP3A4も代謝に関与している。[16.4.1参照]
薬物代謝酵素用語
CYP2C19
薬物代謝酵素用語
CYP2D6
薬物代謝酵素用語
CYP3A4
10.1 併用禁忌
10.2 併用注意
| セロトニン作用薬 トリプタン系薬剤 スマトリプタンコハク酸塩 等 選択的セロトニン再取り込み阻害剤 セロトニン前駆物質(L-トリプトファン)含有製剤又は食品等 トラマドール塩酸塩 リネゾリド 炭酸リチウム セイヨウオトギリソウ(St.John's Wort,セント・ジョーンズ・ワート)含有食品 等 [11.1.3参照] | セロトニン症候群等のセロトニン作用による症状があらわれることがある。これらの薬物を併用する際には観察を十分に行うこと。 | 本剤はセロトニン再取り込み阻害作用を有するため、併用により、セロトニン作用が増強することがある。 |
| メチルチオニニウム塩化物水和物(メチレンブルー) [11.1.3参照] | セロトニン症候群等のセロトニン作用による症状があらわれることがある。これらの薬物を併用する際には観察を十分に行うこと。 | メチルチオニニウム塩化物水和物はMAO阻害作用を有するため、セロトニン作用が増強される。 |
| 三環系抗うつ剤 イミプラミン塩酸塩 クロミプラミン塩酸塩 ノルトリプチリン塩酸塩 等 フェノチアジン系抗精神病剤 リスペリドン ブチロフェノン系抗精神病剤 ハロペリドール 抗不整脈剤 フレカイニド酢酸塩 プロパフェノン塩酸塩 [16.7.1参照] | これらの薬剤の血中濃度が上昇するおそれがあるので、これらの薬剤を減量するなど注意すること。 | 本剤がこれらの薬剤の代謝酵素であるCYP2D6を阻害することによると考えられる。 |
| β遮断剤 メトプロロール酒石酸塩 [16.7.1参照] | メトプロロールの血中濃度が上昇するおそれがあるので、メトプロロールを減量するなど注意すること。 | 本剤がこれらの薬剤の代謝酵素であるCYP2D6を阻害することによると考えられる。 |
| シメチジン [16.7.1参照] | 本剤の血中濃度が上昇するおそれがあるので、本剤を減量するなど注意すること。 | シメチジンが本剤の代謝酵素を阻害することによると考えられる。 |
| オメプラゾール ランソプラゾール チクロピジン塩酸塩 [16.7.1参照] | 本剤の血中濃度が上昇するおそれがあるので、本剤を減量するなど注意すること。 | これらの薬剤が本剤の代謝酵素であるCYP2C19を阻害することによると考えられる。 |
| ワルファリンカリウム [16.7.2参照] | 本剤のラセミ体であるシタロプラムとワルファリンとの併用により、ワルファリンのプロトロンビン時間が軽度延長(約5%)したとの報告がある。 本剤の投与を開始もしくは中止する場合は、プロトロンビン時間を慎重にモニターすること。 | 機序は不明である。 |
| 出血傾向が増強する薬剤 非定型抗精神病剤 フェノチアジン系抗精神病剤 三環系抗うつ剤 アスピリン等の非ステロイド系抗炎症剤 ワルファリンカリウム 等 [9.1.8参照] | 出血傾向が増強することがある。 | SSRIの投与により血小板凝集能が阻害され、これらの薬剤との併用により出血傾向が増強することがある。 |
| アルコール(飲酒) | 本剤服用中は飲酒を避けることが望ましい。 | 他の抗うつ剤で作用の増強が報告されている。 |
| QT延長を起こすことが知られている薬剤 [9.1.1、11.1.4参照] | QT延長を起こすおそれがある。 | 併用によりQT延長作用が相加的に増強するおそれがある。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 痙攣(0.1%)[9.1.7参照]
11.1.2 抗利尿ホルモン不適合分泌症候群(SIADH)(頻度不明)
低ナトリウム血症、頭痛、集中力の欠如、記憶障害、錯乱、幻覚、痙攣、失神等を伴う抗利尿ホルモン不適合分泌症候群(SIADH)があらわれることがあるので、異常が認められた場合には投与を中止し、水分摂取の制限等適切な処置を行うこと。
11.1.3 セロトニン症候群(頻度不明)
11.2 その他の副作用
| 5%以上 | 1〜5%未満 | 1%未満 | 頻度不明 | |
| 全身症状 | 倦怠感 | 異常感 | 無力症、浮腫、熱感、発熱、悪寒、疲労、体重増加、体重減少 | |
| 過敏症 | 発疹、湿疹、蕁麻疹、そう痒 | アナフィラキシー反応、血管浮腫 | ||
| 精神神経系 | 傾眠(22.6%)、浮動性めまい、頭痛 | あくび、不眠症、体位性めまい、感覚鈍麻、易刺激性(いらいら感、焦燥) | アカシジア、睡眠障害、異常夢(悪夢を含む)、激越、不安、錯乱状態、躁病、落ち着きのなさ、錯感覚(ピリピリ感等)、振戦、リビドー減退、歯ぎしり | パニック発作、精神運動不穏、失神、幻覚、神経過敏、離人症、ジスキネジー、運動障害、無オルガズム症 |
| 消化器 | 悪心(20.7%)、口渇 | 腹部不快感、下痢、食欲減退、腹痛、嘔吐、便秘 | 腹部膨満、胃炎、食欲亢進、消化不良 | |
| 循環器 | 動悸 | 起立性低血圧、QT延長 | 頻脈、徐脈 | |
| 血液 | 赤血球減少、ヘマトクリット減少、ヘモグロビン減少、白血球増加、血小板増加、血小板減少、鼻出血 | 出血傾向(斑状出血、消化管出血等) | ||
| 肝臓 | AST・ALT・Al-P・γ-GTP・ビリルビンの上昇等の肝機能検査値異常 | 肝炎 | ||
| 筋骨格系 | 関節痛、筋肉痛、肩こり、こわばり | |||
| 泌尿器・生殖器 | 排尿困難、尿蛋白陽性、射精障害 | 頻尿、尿閉、不正出血、勃起不全、射精遅延 | 持続勃起症、月経過多 | |
| その他 | 回転性めまい、耳鳴、多汗症 | 副鼻腔炎、味覚異常、脱毛、コレステロール上昇、血中ナトリウム低下、乳汁漏出、胸部不快感、寝汗、羞明、霧視、過換気、尿糖陽性 | 視覚異常、散瞳、高プロラクチン血症 |
13. 過量投与
13.1 症状
海外において、本剤1000mgを超える過量投与が報告されている。また、本剤を過量投与した患者において、死亡例が海外で報告されている。主な症状として、中枢神経障害(めまい、振戦、不安、焦燥、興奮、セロトニン症候群、痙攣、昏睡)、胃腸障害(悪心・嘔吐等)、心血管障害(低血圧、頻脈、QT延長、不整脈)、電解質及び水分バランス異常(低カリウム血症、低ナトリウム血症)等が報告されている。
13.2 処置
特異的な解毒剤は知られていない。
14. 適用上の注意
14.1 薬剤交付時の注意
<製剤共通>
14.1.1 PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
<OD錠>
14.1.2 本剤は舌の上にのせて唾液を浸潤させると崩壊するため、水なしで服用可能である。また、水で服用することもできる。
14.1.3 本剤は寝たままの状態では、水なしで服用させないこと。
15. その他の注意
15.1 臨床使用に基づく情報
15.1.1 海外で実施された大うつ病性障害等の精神疾患を有する患者を対象とした、本剤を含む複数の抗うつ剤の短期プラセボ対照臨床試験の検討結果において、24歳以下の患者では、自殺念慮や自殺企図の発現のリスクが抗うつ剤投与群でプラセボ群と比較して高かった。なお、25歳以上の患者における自殺念慮や自殺企図の発現のリスクの上昇は認められず、65歳以上においてはそのリスクが減少した。[5.1、8.1-8.4、9.1.3、9.1.4参照]
15.1.2 主に50歳以上を対象に実施された海外の疫学調査において、選択的セロトニン再取り込み阻害剤及び三環系抗うつ剤を含む抗うつ剤を投与された患者で、骨折のリスクが上昇したとの報告がある。
15.1.3 海外で実施された臨床試験において、本剤を含む選択的セロトニン再取り込み阻害剤が精子特性を変化させ、受精率に影響を与える可能性が報告されている。
15.2 非臨床試験に基づく情報
15.2.1 ラット反復投与毒性試験において、本剤投与後に、心毒性(心筋炎に基づくうっ血性心不全)による死亡が認められている。心毒性は本剤のCmaxに依存して発現するものと考えられ、発現の閾値におけるラット及びヒト曝露量の乖離は約8倍と推察されている。
15.2.2 ラット反復投与毒性試験において、本剤投与後に、肺、精巣上体及び副腎にリン脂質症に関連する所見(光顕的に認められる泡沫状肺胞マクロファージの集簇及び細胞の空胞化)が認められ、これらの所見はヒトにおける曝露量よりも低い曝露量より認められた。休薬により、リン脂質症に関連する所見は回復した。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
健康成人(CYP2C19のPM(Poor Metabolizer)及びEM(Extensive Metabolizer)各6例)に絶食下でエスシタロプラム5mg、10mg、20mgを単回経口投与した。CYP2C19EM群では投与後3.8〜4.3時間で最高血漿中濃度(Cmax)に達し、消失半減期(t1/2)は24.6〜27.7時間であり、Cmax及び血中濃度−時間曲線下面積(AUC)は投与量にほぼ比例して増加した。CYP2C19PM群における最高血漿中濃度到達時間及びCmaxはCYP2C19EM群と同程度であったが、AUC及びt1/2はCYP2C19EM群の約2倍であった。4)[7.2、9.1.2、16.6.4参照]
| CYP2C19遺伝子型注1) | 投与量(mg) | Cmax(ng/mL) | tmax(hr) | AUC0-∞(ng・hr/mL) | t1/2(hr) |
| EM | 5 | 5.7±0.8 | 3.8±1.3 | 183±70 | 24.6±9.9 |
| 10 | 10.8±2.1 | 3.8±0.4 | 418±153 | 27.7±7.5 | |
| 20 | 23.0±4.3 | 4.3±1.4 | 807±282 | 27.4±7.2 | |
| PM | 5 | 5.5±0.6 | 4.2±1.5 | 384±109 | 55.8±16.4 |
| 10 | 12.9±2.3 | 4.8±1.8 | 885±384 | 51.2±16.9 | |
| 20 | 24.7±4.7 | 5.2±1.8 | 1595±356 | 55.3±8.7 |
16.1.2 反復投与
健康成人(CYP2C19のPM及びEM各5例)にエスシタロプラム10mgを1日1回21日間反復経口投与した。CYP2C19EM群、CYP2C19PM群のいずれにおいても血漿中濃度は投与回数に従い徐々に上昇し、CYP2C19EM群では投与15日目までに、CYP2C19PM群では投与19日目までにほぼ定常状態に達した。CYP2C19PM群の21日間反復投与後におけるCmax、AUC及びt1/2のいずれも、CYP2C19EM群と比較して約2倍高値であった。4)[7.2、9.1.2、16.6.4参照]
| CYP2C19遺伝子型注2) | Cmax(ng/mL) | tmax(hr) | AUC0-24(ng・hr/mL) | t1/2(hr) |
| EM | 26.8±6.1 | 3.0±1.0 | 506±132 | 37.7±7.5 |
| PM | 53.9±12.9 | 6.4±3.3 | 1094±266 | 57.8±14.7 |
16.1.3 生物学的同等性試験
<エスシタロプラム錠10mg「トーワ」>
エスシタロプラム錠10mg「トーワ」とレクサプロ錠10mgを、クロスオーバー法によりそれぞれ1錠(エスシタロプラムとして10mg)健康成人男子に絶食単回経口投与して血漿中未変化体濃度を測定し、得られた薬物動態パラメータ(AUC、Cmax)について90%信頼区間法にて統計解析を行った結果、いずれもlog(0.80)〜log(1.25)の範囲内であり、両剤の生物学的同等性が確認された。5)
| 判定パラメータ | 参考パラメータ | |||
| AUC0-72(ng・hr/mL) | Cmax(ng/mL) | tmax(hr) | t1/2(hr) | |
| エスシタロプラム錠10mg「トーワ」 | 241.1±61.9 | 10.00±2.30 | 3.2±1.3 | 22.88±4.87 |
| レクサプロ錠10mg | 242.3±58.8 | 9.68±2.10 | 3.5±1.1 | 22.84±5.57 |
血漿中濃度並びにAUC、Cmax等のパラメータは、被験者の選択、体液の採取回数・時間等の試験条件によって異なる可能性がある。
<エスシタロプラムOD錠10mg「トーワ」>
エスシタロプラムOD錠10mg「トーワ」とレクサプロ錠10mgを、クロスオーバー法によりそれぞれ1錠(エスシタロプラムとして10mg)健康成人男子に絶食単回経口投与(水なしで服用及び水で服用)して血漿中未変化体濃度を測定し、得られた薬物動態パラメータ(AUC、Cmax)について90%信頼区間法にて統計解析を行った結果、いずれもlog(0.80)〜log(1.25)の範囲内であり、両剤の生物学的同等性が確認された。6)
(1)水なしで服用(レクサプロ錠は水で服用)
| 判定パラメータ | 参考パラメータ | |||
| AUC0-72(ng・hr/mL) | Cmax(ng/mL) | tmax(hr) | t1/2(hr) | |
| エスシタロプラムOD錠10mg「トーワ」 | 243.5±41.0 | 10.99±2.05 | 3.0±0.7 | 23.41±4.37 |
| レクサプロ錠10mg | 246.5±44.1 | 11.41±2.12 | 2.5±0.5 | 23.80±4.19 |
血漿中濃度並びにAUC、Cmax等のパラメータは、被験者の選択、体液の採取回数・時間等の試験条件によって異なる可能性がある。
(2)水で服用
| 判定パラメータ | 参考パラメータ | |||
| AUC0-72(ng・hr/mL) | Cmax(ng/mL) | tmax(hr) | t1/2(hr) | |
| エスシタロプラムOD錠10mg「トーワ」 | 268.8±78.4 | 13.13±3.67 | 2.4±0.5 | 23.62±4.01 |
| レクサプロ錠10mg | 275.8±82.4 | 13.41±3.60 | 2.5±1.0 | 23.61±4.44 |
血漿中濃度並びにAUC、Cmax等のパラメータは、被験者の選択、体液の採取回数・時間等の試験条件によって異なる可能性がある。
16.2 吸収
16.2.1 食事の影響
健康成人(17例)に絶食下又は高脂肪食摂取後にエスシタロプラム20mgを単回経口投与したとき、Cmax及びAUCは両群で統計学的有意差は認められず、食事の影響は認められなかった(外国人データ)。7)
16.2.2 生物学的利用率
エスシタロプラムのラセミ体であるシタロプラム40mgを健康成人12例に単回経口投与したときの生物学的利用率は79.5%であった(外国人データ)。8)
16.3 分布
16.3.1 分布容積
健康成人(CYP2C19のPM及びEM各6例)にエスシタロプラム5mg、10mg、20mgを単回経口投与したときのみかけの分布容積(Vz/F)は872〜1053Lであった。4)
16.3.2 血漿蛋白結合率
ヒト血漿にエスシタロプラム(20〜100ng/mL)を添加したとき、検討した濃度範囲における血漿蛋白結合率はほぼ一定であり、その平均値は55.4%であった(in vitro、外国人データ)。9)
16.4 代謝
16.4.1 エスシタロプラムは主にCYP2C19によりデメチル化体へ代謝され、また、デメチル化体への代謝には、CYP2D6及びCYP3A4が関与する。デメチル化体はCYP2D6によりジデメチル化体へ代謝される。また、エスシタロプラムの一部はCYP2D6あるいはモノアミンオキシダーゼ並びにアルデヒド酸化酵素により酸化されN-オキサイド体あるいはプロピオン酸体に代謝されることが報告されている。エスシタロプラムは、肝臓でこれら代謝物に変換された後、そのまま、あるいはグルクロン酸抱合体として尿中に排泄されると考えられる。10)11)12)13)[10.参照]
16.4.2 健康成人にエスシタロプラムを単回あるいは反復経口投与したときのCmax及びAUCは、エスシタロプラム、デメチル化体、ジデメチル化体の順に高かった。また、ジデメチル化体の尿中排泄率は、エスシタロプラムあるいはデメチル化体に比較して低かった。4)
16.5 排泄
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
16.6.2 肝機能障害患者
16.6.3 高齢者
16.6.4 薬物代謝酵素の活性が遺伝的に欠損している者
(2)CYP2D6
エスシタロプラムを健康成人に経口投与あるいは静脈内投与注3)したとき、CYP2D6PMにおけるCmax及びAUCは、8例中1例でCYP2D6EMにおける値のそれぞれ1.2倍及び1.3倍であったが、他の7例ではCYP2D6EMと同程度であった(外国人データ)。18)
注3)本剤の承認用法及び用量は、1日1回20mgまでの経口投与である。
16.7 薬物相互作用
16.7.1 エスシタロプラムを用いた試験の成績
(1)オメプラゾール
(2)シメチジン
(3)メトプロロール
(4)デシプラミン
(5)リトナビル
健康成人(18例)にエスシタロプラム20mgとリトナビル600mgを併用経口投与したとき、エスシタロプラム及びリトナビルの薬物動態に影響は認められなかった(外国人データ)。19)
16.7.2 エスシタロプラムのラセミ体であるシタロプラム(国内未発売)を用いた試験の成績
(1)レボメプロマジン
健康成人(8例)にシタロプラム40mgを1日1回10日間反復経口投与し、7日目にレボメプロマジン50mgを併用経口投与したとき、シタロプラム及びレボメプロマジンの薬物動態に影響は認められなかった(外国人データ)。19)
(2)トリアゾラム
健康成人(17例)にシタロプラムを反復経口投与(20mg/日を7日間、引き続き40mg/日を23日間)し、最終投与日(30日)にトリアゾラム0.25mgを併用経口投与したとき、シタロプラム及びトリアゾラムの薬物動態に影響は認められなかった(外国人データ)。19)
(3)カルバマゼピン
健康成人(12例)にカルバマゼピンを反復経口投与(100mgを2回/日を3日間、引き続き200mgを2回/日を3日間、400mg/日を29日間)し、22日目よりシタロプラム40mgを1日1回14日間反復併用経口投与したとき、カルバマゼピンの薬物動態に影響は認められなかった(外国人データ)。19)
(4)ピモジド
(5)ケトコナゾール
健康成人(17例)にシタロプラム40mg及びケトコナゾール(経口剤は国内未発売)200mgを併用経口投与したとき、シタロプラムの薬物動態に影響は認められなかった。また、ケトコナゾール単独投与時と比べ、ケトコナゾールのtmaxは遅延(併用時2.4時間、単独投与時1.9時間)し、Cmaxが0.79倍に低下したが、AUC及びt1/2は同程度であった(外国人データ)。19)
(6)ワルファリン
(7)ジゴキシン
健康成人(11例)にシタロプラム40mgを1日1回29日間反復経口投与し、22日目にジゴキシン1mgを併用経口投与したとき、シタロプラム及びジゴキシンの薬物動態に影響は認められなかった(外国人データ)。19)
(8)リチウム
健康成人(8例)にシタロプラム40mgを1日1回10日間反復経口投与し、3日目から7日目までリチウム30mmolを1日1回5日間反復併用経口投与したとき、シタロプラム及びリチウムの薬物動態に影響は認められなかった(外国人データ)。19)
16.8 その他
17. 臨床成績
17.1 有効性及び安全性に関する試験
<うつ病・うつ状態>
17.1.1 国内第III相試験
大うつ病性障害患者を対象として、エスシタロプラムシュウ酸塩(エスシタロプラムとして1日10mg又は20mg)、プラセボ又はパロキセチン塩酸塩水和物(パロキセチンとして1日20〜40mg)を8週間投与した結果、主要評価項目であるMontgomery Asberg Depression Rating Scale(MADRS)合計点の変化量は下表のとおりであり、エスシタロプラム(10mg及び20mg併合群)のプラセボに対する優越性が示された。
観察期及び後観察期の副作用発現頻度は、エスシタロプラム10mg投与群で63.3%(76/120例)、エスシタロプラム20mg投与群で75.6%(90/119例)であった。主な副作用は、10mg投与群では傾眠15.0%(18/120例)、悪心13.3%(16/120例)、浮動性めまい9.2%(11/120例)、20mg投与群では傾眠20.2%(24/119例)、悪心21.0%(25/119例)、浮動性めまい10.1%(12/119例)であった。22)
観察期及び後観察期の副作用発現頻度は、エスシタロプラム10mg投与群で63.3%(76/120例)、エスシタロプラム20mg投与群で75.6%(90/119例)であった。主な副作用は、10mg投与群では傾眠15.0%(18/120例)、悪心13.3%(16/120例)、浮動性めまい9.2%(11/120例)、20mg投与群では傾眠20.2%(24/119例)、悪心21.0%(25/119例)、浮動性めまい10.1%(12/119例)であった。22)
| 投与群 | 例数 | MADRS合計点注1) | 変化量 | ||||
| ベースライン | 最終評価時 | ベースラインからの変化量注1) | プラセボ群との対比較注2) | ||||
| 群間差注3)[95%信頼区間] | p値 | ||||||
| プラセボ群 | 124 | 29.0±5.6 | 18.3±10.1 | -10.7±9.5 | − | − | |
| エスシタロプラム | 10mg群 | 120 | 29.4±5.8 | 15.6±11.0 | -13.7±10.0 | -3.0[-5.4,-0.5] | 0.018注4) |
| 20mg群 | 119 | 29.8±6.0 | 16.2±10.1 | -13.6±8.8 | -2.7[-5.0,-0.4] | 0.021注4) | |
| 併合群 | 239 | 29.6±5.9 | 15.9±10.5 | -13.7±9.4 | -2.8[-4.9,-0.8] | 0.006注4) | |
| パロキセチン群 | 121 | 29.8±5.9 | 15.6±10.0 | -14.2±9.9 | -3.2[-5.6,-0.8] | 0.009注4) | |
17.1.2 国内第III相長期投与試験
大うつ病性障害患者を対象として、エスシタロプラムシュウ酸塩(エスシタロプラムとして1日10mg又は20mg)を最大52週間投与した結果、52週まで有効性は維持された。
観察期及び後観察期の副作用発現頻度は、エスシタロプラム10mg又は20mg投与群では80.4%(74/92例)であった。主な副作用は、傾眠30.4%(28/92例)、悪心23.9%(22/92例)、頭痛19.6%(18/92例)、浮動性めまい15.2%(14/92例)であった。23)
観察期及び後観察期の副作用発現頻度は、エスシタロプラム10mg又は20mg投与群では80.4%(74/92例)であった。主な副作用は、傾眠30.4%(28/92例)、悪心23.9%(22/92例)、頭痛19.6%(18/92例)、浮動性めまい15.2%(14/92例)であった。23)
| 評価時期 | 例数 | MADRS合計点注5) | 変化量注5) |
| ベースライン | 92 | 31.3±5.5 | − |
| 8週時 | 87 | 15.0±9.3 | -16.5±8.5 |
| 24週時 | 79 | 10.8±9.1 | -20.3±8.6 |
| 52週時 | 66 | 8.0±7.4 | -23.0±7.6 |
17.1.3 国内第III相高齢者長期投与試験
高齢の大うつ病性障害患者を対象として、エスシタロプラムシュウ酸塩(エスシタロプラムとして1日10mg又は20mg)を最大52週間投与した結果、52週まで有効性は維持された。
観察期及び後観察期の副作用発現頻度は、エスシタロプラム10mg又は20mg投与群では81.8%(18/22例)であった。主な副作用は、口渇、傾眠及び悪心各22.7%(5/22例)であった。24)
観察期及び後観察期の副作用発現頻度は、エスシタロプラム10mg又は20mg投与群では81.8%(18/22例)であった。主な副作用は、口渇、傾眠及び悪心各22.7%(5/22例)であった。24)
| 評価時期 | 例数 | MADRS合計点注6) | 変化量注6) |
| ベースライン | 22 | 31.4±8.6 | − |
| 8週時 | 19 | 17.1±9.9 | -13.7±9.0 |
| 24週時 | 14 | 11.5±8.5 | -18.6±7.6 |
| 52週時 | 13 | 7.4±6.4 | -23.3±6.6 |
<社会不安障害>
17.1.4 国内第III相試験
社会不安障害患者を対象として、エスシタロプラムシュウ酸塩(エスシタロプラムとして1日10mg又は20mg)又はプラセボを12週間投与した結果、主要評価項目であるLiebowitz Social Anxiety Scale-J(LSAS-J)合計点の変化量は下表のとおりであった。
観察期及び後観察期の副作用発現頻度は、エスシタロプラム10mg投与群で51.5%(102/198例)、エスシタロプラム20mg投与群で57.5%(111/193例)であった。主な副作用は、10mg群では傾眠18.7%(37/198例)、悪心14.6%(29/198例)、20mg投与群では傾眠22.3%(43/193例)、悪心17.6%(34/193例)であった。25)
観察期及び後観察期の副作用発現頻度は、エスシタロプラム10mg投与群で51.5%(102/198例)、エスシタロプラム20mg投与群で57.5%(111/193例)であった。主な副作用は、10mg群では傾眠18.7%(37/198例)、悪心14.6%(29/198例)、20mg投与群では傾眠22.3%(43/193例)、悪心17.6%(34/193例)であった。25)
| 投与群 | 例数 | LSAS-J合計点注7) | 変化量 | ||||
| ベースライン | 投与12週時 | ベースラインからの変化量注7) | プラセボ群との対比較注8) | ||||
| 群間差注9)[95%信頼区間] | p値 | ||||||
| プラセボ群 | 196 | 95.3±18.5 | 72.2±27.4 | -23.1±21.4 | − | − | |
| エスシタロプラム | 10mg群 | 198 | 94.5±18.2 | 67.6±29.0 | -26.9±23.3 | -3.9[-8.3,0.6] | 0.089 |
| 20mg群 | 193 | 93.4±17.8 | 60.7±28.0 | -32.6±25.6 | -9.8[-14.5,-5.2] | −注10) | |
17.1.5 国内第III相長期投与試験
社会不安障害患者を対象として、エスシタロプラムシュウ酸塩(エスシタロプラムとして1日10mg又は20mg)を最大52週間投与した結果、52週まで有効性は維持された。
観察期及び後観察期の副作用発現頻度は、エスシタロプラム10mg又は20mg投与群では60.1%(95/158例)であった。観察期の主な副作用は、傾眠24.7%(39/158例)、悪心19.0%(30/158例)であった。後観察期において発現率が10%以上の副作用は認められなかった。26)
観察期及び後観察期の副作用発現頻度は、エスシタロプラム10mg又は20mg投与群では60.1%(95/158例)であった。観察期の主な副作用は、傾眠24.7%(39/158例)、悪心19.0%(30/158例)であった。後観察期において発現率が10%以上の副作用は認められなかった。26)
| 評価時期 | 例数 | LSAS-J合計点注11) | 変化量注11) |
| ベースライン | 158 | 95.3±19.5 | − |
| 12週時 | 141 | 69.0±25.1 | -26.6±21.5 |
| 24週時 | 138 | 59.9±28.7 | -35.6±27.2 |
| 52週時 | 126 | 49.9±28.0 | -44.8±28.8 |
17.3 その他
17.3.1 QT間隔に対する影響
健康成人117例を対象としたプラセボ対照二重盲検比較試験(Thorough QT試験)において、QTcFのベースラインからの変化量(プラセボ補正)は、エスシタロプラム1日10mg投与において4.3msec、1日30mg投与注12)において10.7msecであった(外国人データ)。27)
| 薬剤 | QTcF(90%信頼区間)(msec) |
| エスシタロプラム 10mg/日 | 4.3(2.2,6.4) |
| エスシタロプラム 30mg/日注12) | 10.7(8.6,12.8) |
| モキシフロキサシン 400mg/日 | 9.2(7.7,10.7) |
注12)本剤の承認用法及び用量は、1日1回20mgまでの経口投与である。
18. 薬効薬理
18.1 作用機序
エスシタロプラムは選択的なセロトニン(5-HT)再取り込み阻害作用を示し、脳内での細胞外5-HT濃度を持続的に上昇させることにより5-HT神経系を賦活化し抗うつ作用を示すと考えられる。28)
18.2 抗うつ作用
18.2.3 ラット社会的ストレスモデルにおいて、居住ラットの侵入ラットに対する攻撃行動を単回投与では減少させ、逆に反復投与では増加させた。33)
18.3 セロトニン再取り込み阻害作用
18.3.1 ラット脳シナプトソームを用いたin vitro実験において5-HT取り込みを阻害し(50%抑制濃度は2.1nmol/L)、in vivoにおいてもラット前頭皮質中の細胞外5-HT濃度を上昇させた。34)35)
18.3.2 ヒトモノアミントランスポータ発現細胞において、エスシタロプラムの5-HTトランスポータに対する選択性(結合親和性定数の比率)はノルアドレナリントランスポータ及びドパミントランスポータと比較して各々7100倍及び24000倍であった(in vitro)。36)
19. 有効成分に関する理化学的知見
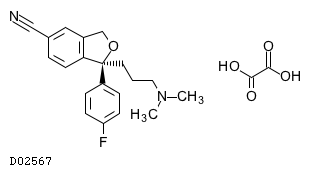
19.1. エスシタロプラムシュウ酸塩
| 一般的名称 | エスシタロプラムシュウ酸塩 |
|---|---|
| 一般的名称(欧名) | Escitalopram Oxalate |
| 化学名 | (1S)-1-[3-(Dimethylamino)propyl]-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitrile monooxalate |
| 分子式 | C20H21FN2O・C2H2O4 |
| 分子量 | 414.43 |
| 物理化学的性状 | 白色〜淡黄色の粉末である。酢酸(100)に極めて溶けやすく、メタノール又はN,N-ジメチルアセトアミドに溶けやすく、水にやや溶けやすく、アセトニトリルにやや溶けにくく、エタノール(99.5)に溶けにくい。 |
| KEGG DRUG |
|
22. 包装
<エスシタロプラム錠10mg「トーワ」>
100錠[10錠×10:PTP]
500錠[10錠×50:PTP]
300錠[バラ]
<エスシタロプラム錠20mg「トーワ」>
100錠[10錠×10:PTP]
<エスシタロプラムOD錠10mg「トーワ」>
100錠[10錠×10:PTP]
300錠[バラ、乾燥剤入り]
<エスシタロプラムOD錠20mg「トーワ」>
100錠[10錠×10:PTP]
23. 主要文献
- Chambers,C.D.et al., N.Engl.J.Med., 354 (6), 579-587, (2006) »PubMed
- Kallen,B.et al, Pharmacoepidemiol.Drug Saf., 17 (8), 801-806, (2008) »PubMed
- Wagner,K.D.et al., J.Am.Acad.Child Adolesc.Psychiatry., 45 (3), 280-288, (2006) »PubMed
- 国内第I相試験−エスシタロプラムの単回及び反復投与試験−(レクサプロ錠:2011年4月22日承認、申請資料概要2.7.6.4)
- 社内資料:生物学的同等性試験(錠10mg)
- 吉原達也 他, 医学と薬学, 79 (10), 1325-1337, (2022)
- 海外臨床薬物動態試験−エスシタロプラムの薬物動態に及ぼす食事の影響−(レクサプロ錠:2011年4月22日承認、申請資料概要2.7.6.3)
- 海外臨床薬物動態試験−シタロプラムの生物学的利用率−(レクサプロ錠:2011年4月22日承認、申請資料概要2.7.6.2)
- 薬物動態試験−エスシタロプラムの蛋白結合の検討−(レクサプロ錠:2011年4月22日承認、申請資料概要2.6.4.4、2.6.5.6)
- 薬物動態試験−エスシタロプラムのin vitro代謝の検討−(レクサプロ錠:2011年4月22日承認、申請資料概要2.6.4.5)
- Olesen,O.V.et al., Pharmacology., 59 (6), 298-309, (1999) »PubMed
- Rochat,B.et al., Biochem.Pharmacol., 56 (1), 15-23, (1998) »PubMed
- 海外臨床薬物動態試験−シタロプラムのマスバランス−(レクサプロ錠:2011年4月22日承認、申請資料概要2.7.6.4)
- 海外臨床薬物動態試験−腎機能障害患者におけるシタロプラムの薬物動態−(レクサプロ錠:2011年4月22日承認、申請資料概要2.7.6.5)
- 海外臨床薬物動態試験−肝機能障害患者におけるエスシタロプラムの薬物動態−(レクサプロ錠:2011年4月22日承認、申請資料概要2.7.6.5)
- 海外臨床薬物動態試験−高齢者におけるエスシタロプラムの薬物動態(単回投与)−(レクサプロ錠:2011年4月22日承認、申請資料概要2.7.6.5)
- 海外臨床薬物動態試験−高齢者におけるエスシタロプラムの薬物動態(反復投与)−(レクサプロ錠:2011年4月22日承認、申請資料概要2.7.6.5)
- 海外臨床薬物動態試験−エスシタロプラムの薬物動態に及ぼすCYP2D6遺伝子多型の影響−(レクサプロ錠:2011年4月22日承認、申請資料概要2.7.2.3)
- 海外臨床薬物動態試験−エスシタロプラム及びシタロプラムの薬物相互作用試験−(レクサプロ錠:2011年4月22日承認、申請資料概要2.7.6.6)
- 社内資料:生物学的同等性試験(錠20mg)
- 社内資料:生物学的同等性試験(OD錠20mg)
- 用量反応非劣性試験−大うつ病性障害患者におけるプラセボ及び塩酸パロキセチンを対照とした有効性及び安全性の検討−(レクサプロ錠:2011年4月22日承認、申請資料概要2.7.3.2、2.7.6.8)
- 長期投与試験−大うつ病性障害患者における長期投与の安全性及び有効性の検討−(レクサプロ錠:2011年4月22日承認、申請資料概要2.7.3.2、2.7.6.9)
- 高齢者長期投与試験−大うつ病性障害患者における長期投与の安全性、有効性及び薬物動態の検討−(レクサプロ錠:2011年4月22日承認、申請資料概要2.7.3.2、2.7.6.9)
- 社会不安障害に対するプラセボ対照試験(レクサプロ錠:2015年11月20日承認、申請資料概要2.7.6.1)
- 社会不安障害に対する長期投与試験(レクサプロ錠:2015年11月20日承認、申請資料概要2.7.6.2)
- 海外Thorough QT試験−エスシタロプラムの心臓再分極に及ぼす影響−(レクサプロ錠:2011年4月22日承認、申請資料概要2.7.6.7)
- 作用機序(レクサプロ錠:2011年4月22日承認、申請資料概要2.6.2.2)
- 薬理試験−うつ病モデルにおける有効性−(レクサプロ錠:2011年4月22日承認、申請資料概要2.6.2.2)
- Sanchez,C.et al., Psychopharmacology., 167 (4), 353-362, (2003) »PubMed
- Montgomery,S.A.et al., Pharmacol.Toxicol., 88 (5), 282-286, (2001) »PubMed
- Sanchez,C.et al., Behav.Pharmacol., 14 (5-6), 465-470, (2003) »PubMed
- 薬理試験−ラット社会的ストレスモデルの行動様式に及ぼす影響−(レクサプロ錠:2011年4月22日承認、申請資料概要2.6.2.2)
- 薬理試験−ラット脳シナプトソームの5-HT取り込み(in vitro)及びテトラベナジン誘発によるマウスの行動(in vivo)に及ぼす影響−(レクサプロ錠:2011年4月22日承認、申請資料概要2.6.2.2、2.6.2.3)
- Mork,A.et al., Neuropharmacology., 45 (2), 167-173, (2003) »PubMed
- Owens,M.J.et al., Biol.Psychiatry., 50 (5), 345-350, (2001) »PubMed
- Hyttel,J.et al., J.Neural Transm.Gen.Sect., 88 (2), 157-160, (1992) »PubMed
- 薬理試験−エスシタロプラム及び代謝物のモノアミン取り込みに及ぼす影響(in vitro及びin vivo)−(レクサプロ錠:2011年4月22日承認、申請資料概要2.6.2.2、2.6.2.3)
- 薬理試験−各種受容体及びトランスポータに対するリガンドの結合に及ぼす影響−(レクサプロ錠:2011年4月22日承認、申請資料概要2.6.2.2、2.6.2.3)
24. 文献請求先及び問い合わせ先
文献請求先
東和薬品株式会社
学術部DIセンター
〒570-0081
大阪府守口市日吉町2丁目5番15号
電話:0120-108-932
FAX:06-7177-7379
製品情報問い合わせ先
東和薬品株式会社
学術部DIセンター
〒570-0081
大阪府守口市日吉町2丁目5番15号
電話:0120-108-932
FAX:06-7177-7379
26. 製造販売業者等
26.1 製造販売元
東和薬品株式会社
大阪府門真市新橋町2番11号