医薬品情報
| 総称名 | マンジャロ |
|---|---|
| 一般名 | チルゼパチド |
| 欧文一般名 | Tirzepatide |
| 製剤名 | チルゼパチド注射液 |
| 薬効分類名 | 持続性GIP/GLP-1受容体作動薬 |
| 薬効分類番号 | 2499 |
| ATCコード | A10BX16 |
| KEGG DRUG |
D11360
チルゼパチド
|
| KEGG DGROUP |
DG01493
GLP-1受容体作動薬
DG02044
血糖降下薬
|
| JAPIC | 添付文書(PDF) |
添付文書情報2026年5月 改訂(第10版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| マンジャロ皮下注2.5mgアテオス | Mounjaro Subcutaneous Injection ATEOS | 日本イーライリリー | 2499422G1024 | 1924円/キット | 劇薬, 処方箋医薬品 |
| マンジャロ皮下注5mgアテオス | Mounjaro Subcutaneous Injection ATEOS | 日本イーライリリー | 2499422G2020 | 3848円/キット | 劇薬, 処方箋医薬品 |
| マンジャロ皮下注7.5mgアテオス | Mounjaro Subcutaneous Injection ATEOS | 日本イーライリリー | 2499422G3027 | 5772円/キット | 劇薬, 処方箋医薬品 |
| マンジャロ皮下注10mgアテオス | Mounjaro Subcutaneous Injection ATEOS | 日本イーライリリー | 2499422G4023 | 7696円/キット | 劇薬, 処方箋医薬品 |
| マンジャロ皮下注12.5mgアテオス | Mounjaro Subcutaneous Injection ATEOS | 日本イーライリリー | 2499422G5020 | 9620円/キット | 劇薬, 処方箋医薬品 |
| マンジャロ皮下注15mgアテオス | Mounjaro Subcutaneous Injection ATEOS | 日本イーライリリー | 2499422G6026 | 11544円/キット | 劇薬, 処方箋医薬品 |
2. 禁忌
次の患者には投与しないこと
2.1 本剤の成分に対し過敏症の既往歴のある患者
2.2 糖尿病性ケトアシドーシス、糖尿病性昏睡又は前昏睡、1型糖尿病の患者[インスリン製剤による速やかな治療が必須となるので、本剤を投与すべきでない。]
2.3 重症感染症、手術等の緊急の場合[インスリン製剤による血糖管理が望まれるので、本剤の投与は適さない。]
4. 効能または効果
5. 効能または効果に関連する注意
本剤の適用は、あらかじめ糖尿病治療の基本である食事療法、運動療法を十分に行った上で効果が不十分な場合に限り考慮すること。
6. 用法及び用量
通常、成人には、チルゼパチドとして週1回5mgを維持用量とし、皮下注射する。ただし、週1回2.5mgから開始し、4週間投与した後、週1回5mgに増量する。
なお、患者の状態に応じて適宜増減するが、週1回5mgで効果不十分な場合は、4週間以上の間隔で2.5mgずつ増量できる。ただし、最大用量は週1回15mgまでとする。
なお、患者の状態に応じて適宜増減するが、週1回5mgで効果不十分な場合は、4週間以上の間隔で2.5mgずつ増量できる。ただし、最大用量は週1回15mgまでとする。
7. 用法及び用量に関連する注意
7.1 本剤は週1回投与する薬剤であり、同一曜日に投与させること。
7.2 投与を忘れた場合は、次回投与までの期間が3日間(72時間)以上であれば、気づいた時点で直ちに投与し、その後はあらかじめ定めた曜日に投与すること。次回投与までの期間が3日間(72時間)未満であれば投与せず、次のあらかじめ定めた曜日に投与すること。なお、週1回投与の曜日を変更する必要がある場合は、前回投与から少なくとも3日間(72時間)以上間隔を空けること。
7.3 胃腸障害等の発現により忍容性が得られない患者では減量又は漸増の延期を考慮すること。
7.4 本剤投与による用量依存的な体重減少が認められているため、血糖コントロールだけでなく、体重減少にも注意し、本剤の増量の必要性を慎重に判断すること。[9.8参照]
8. 重要な基本的注意
8.1 本剤はインスリンの代替薬ではない。本剤の投与に際しては、患者のインスリン依存状態を確認し、投与の可否を判断すること。インスリン依存状態の患者で、インスリンからGLP-1受容体作動薬に切り替え、急激な高血糖及び糖尿病性ケトアシドーシスが発現した症例が報告されている。
8.2 投与する場合には、血糖、尿糖を定期的に検査し、薬剤の効果を確かめ、3〜4ヵ月間投与して効果が不十分な場合には、より適切と考えられる治療への変更を考慮すること。
8.3 本剤は持続性製剤であり、本剤中止後も効果が持続する可能性があるため、血糖値の変動や副作用予防、副作用発現時の処置について十分留意すること。[16.1参照]
8.5 低血糖を起こすことがあるので、高所作業、自動車の運転等に従事している患者に投与するときは注意すること。[11.1.1参照]
8.6 急性膵炎が発現することがあるので、急性膵炎の初期症状(嘔吐を伴う持続的な激しい腹痛等)があらわれた場合は、使用を中止し、速やかに医師の診断を受けるよう指導すること。[9.1.2、11.1.2参照]
8.8 本剤投与中は、甲状腺関連の症候の有無を確認し、異常が認められた場合には、専門医を受診するよう指導すること。[15.2参照]
8.9 過度の体重減少がみられた場合は、本剤の減量又は投与中止を考慮すること。投与開始時のBody Mass Index(BMI)が23kg/m2未満の患者での本剤の有効性及び安全性は検討されていない。[9.8参照]
8.10 胆石症、胆嚢炎、胆管炎又は胆汁うっ滞性黄疸が発現するおそれがあるので、腹痛等の腹部症状がみられた場合には、必要に応じて画像検査等による原因精査を考慮するなど、適切に対応すること。[11.1.3参照]
8.11 急激な血糖コントロールの改善に伴い、糖尿病網膜症の顕在化又は増悪があらわれることがあるので、注意すること。[9.1.4参照]
8.12 下痢、嘔吐から脱水を続発し、急性腎障害に至るおそれがあるので、患者の状態に注意すること。
8.13 血圧低下がみられた場合には患者の状態を十分に観察し、異常が認められた場合には適切な処置を行うこと。[17.1.2参照]
8.14 本剤の自己注射にあたっては、患者に十分な教育訓練を実施した後、患者自ら確実に投与できることを確認した上で、医師の管理指導のもと実施すること。また、器具の安全な廃棄方法について指導を徹底すること。添付されている取扱説明書を必ず読むよう指導すること。
8.15 本剤はチルゼパチドを含有しているため、ゼップバウンド等他のチルゼパチド含有製剤と併用しないこと。
8.16 本剤とDPP-4阻害剤はいずれもGLP-1受容体及びGIP受容体を介した血糖降下作用を有している。両剤を併用した際の臨床試験成績はなく、有効性及び安全性は確認されていない。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 重症胃不全麻痺等の重度の胃腸障害のある患者
胃腸障害の症状が悪化するおそれがある。
9.1.3 低血糖を起こすおそれがある以下の患者又は状態
9.1.4 増殖糖尿病網膜症、糖尿病黄斑浮腫、急性期治療を要する非増殖糖尿病網膜症を合併する患者又はこれらの既往歴のある患者[8.11参照]
9.1.5 腹部手術の既往又はイレウスの既往のある患者
腸閉塞を含むイレウスを起こすおそれがある。[11.1.5参照]
9.1.6 全身麻酔又は深い鎮静下の患者
GLP-1受容体作動薬又はGIP/GLP-1受容体作動薬を投与中の患者において、術前の絶食指示を遵守したにもかかわらず、全身麻酔又は深い鎮静下で誤嚥が生じた症例が報告されている。本剤は胃内容排出遅延作用があり、胃内容物残留リスクが高まるおそれがある。[18.2.6参照]
9.4 生殖能を有する者
妊娠する可能性のある女性には、本剤投与中及び最終投与後1ヵ月間において避妊する必要性及び適切な避妊法について説明すること。[9.5参照]
9.5 妊婦
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。ヒト母乳中へ移行することがある。本剤投与によるヒトの哺乳中の児への影響は不明である。[16.3.2参照]
9.7 小児等
小児等を対象とした有効性及び安全性を指標とした臨床試験は実施していない。
9.8 高齢者
10. 相互作用
10.2 併用注意
| 糖尿病用薬 ビグアナイド系薬剤 スルホニルウレア剤 速効型インスリン分泌促進剤 α-グルコシダーゼ阻害剤 チアゾリジン系薬剤 DPP-4阻害剤 インスリン製剤 SGLT2阻害剤等 [11.1.1参照] | 低血糖の発現に注意すること。特にスルホニルウレア剤、速効型インスリン分泌促進剤又はインスリン製剤と併用する場合、低血糖のリスクが増加するおそれがある。これらの薬剤と併用する場合、低血糖のリスクを軽減するため、これらの薬剤の減量を検討すること。 | 血糖降下作用が増強される。 |
| 経口避妊薬 [16.7参照] | 特に投与開始初期又は漸増後初期では併用する経口避妊薬の効果を減弱させるおそれがある。 | 本剤の胃内容物排出遅延作用による。 |
| 治療域の狭い薬剤(経口剤) クマリン系薬剤 ワルファリンカリウム | GLP-1受容体作動薬との併用によりワルファリンのtmaxが遅延したとの報告があり、類薬(エキセナチド)で出血を伴うINR増加が報告されている。治療域の狭い薬剤(経口剤)と併用する場合は、患者の状態を慎重に観察すること。 | 本剤の胃内容物排出遅延作用による。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 低血糖(頻度不明)
11.1.2 急性膵炎(0.1%未満)
11.1.3 胆嚢炎(頻度不明)、胆管炎(0.1%未満)、胆汁うっ滞性黄疸(頻度不明)[8.10参照]
11.1.4 アナフィラキシー、血管性浮腫(いずれも頻度不明)
11.1.5 イレウス(頻度不明)
腸閉塞を含むイレウスを起こすおそれがある。高度の便秘、腹部膨満、持続する腹痛、嘔吐等の異常が認められた場合には投与を中止し、適切な処置を行うこと。[9.1.5参照]
11.2 その他の副作用
| 5%以上 | 1〜5%未満 | 1%未満 | |
| 循環器 | 心拍数増加注)、低血圧、血圧低下 | ||
| 消化器 | 悪心、嘔吐、下痢、便秘、腹痛、消化不良、食欲減退 | 腹部膨満、胃食道逆流性疾患、おくび | 鼓腸 |
| 肝胆道 | 胆石症 | ||
| 眼 | 糖尿病網膜症 | ||
| 注射部位 | 注射部位反応(紅斑、そう痒感、疼痛、腫脹等) | ||
| 免疫系 | 過敏症(湿疹、発疹、そう痒性皮疹等) | ||
| 精神神経系 | 味覚不全、異常感覚 | ||
| 臨床検査 | 膵アミラーゼ増加、リパーゼ増加、体重減少 | ||
| その他 | 疲労 |
14. 適用上の注意
14.1 薬剤投与前の注意
注入器の破損又は異常がないこと、薬液の変色や浮遊物がないことを確認すること。
14.2 薬剤投与時の注意
皮下注射は、腹部、大腿部又は上腕部に行う。同じ部位の中で注射する場合、毎回注射する場所を変更すること。静脈内及び筋肉内に投与しないこと。
15. その他の注意
15.1 臨床使用に基づく情報
国内外の第III相試験7試験(5119例)において、抗薬物抗体が評価可能な5025例のうち、抗チルゼパチド抗体が51.1%(2570例)に、内因性GIP又は内因性GLP-1に対する交差抗体はそれぞれ33.9%(1705例)及び14.2%(716例)に発現した。チルゼパチドのGIP受容体又はGLP-1受容体の活性化に対する中和抗体はそれぞれ1.9%(94例)及び2.1%(107例)に発現した。
15.2 非臨床試験に基づく情報
雌雄ラットを用いた2年間がん原性試験において、本剤を0.15、0.50及び1.5mg/kgの用量(それぞれ最大臨床推奨用量をヒトに皮下投与した際のAUCの0.12、0.36及び1.02倍のAUCをもたらす用量)で週2回皮下投与したところ、対照群と比較して、甲状腺C細胞腫瘍(腺腫及び癌)の発生頻度の増加がすべての用量でみられた。rasH2トランスジェニックマウスを用いた6ヵ月間がん原性試験において、本剤を1、3及び10mg/kgの用量で週2回皮下投与したところ、甲状腺C細胞の過形成あるいは腫瘍の発生頻度に増加は認められなかった1)。甲状腺髄様癌の既往のある患者及び甲状腺髄様癌又は多発性内分泌腫瘍症2型の家族歴のある患者に対する本剤の安全性は確立していない。[8.8参照]
16. 薬物動態
16.1 血中濃度
日本人2型糖尿病患者29例に本剤5mg、10mg又は15mgを週1回皮下投与(いずれの用量においても週1回2.5mgで投与を開始し、以後4週間ごとに2.5mgずつ増量)したとき、32週目投与後の薬物動態を評価した。本剤32週目投与後のtmaxの中央値は約24時間、半減期(t1/2)は約5〜6日であり、Cmax及びAUC(0-168)の幾何平均値は概ね用量比例的に増加した。[8.3参照]
薬物動態パラメータ及び血漿中濃度推移を以下に示す2)。
| 投与量 (例数) | tmax(h)注1) | t1/2(h)注2) | Cmax(ng/mL) | AUC(0-168)(ng・h/mL) | CL/F(L/h) | Vz/F(L) |
| 5mg (N=7) | 24.63 (8.00-48.00) | 146 (121-269)注3) | 835 (23) | 94800 (16)注4) | 0.0528 (16)注4) | 11.1 (51)注3) |
| 10mg (N=10) | 23.57 (8.00-72.00) | 121 (104-156)注4) | 1730 (46) | 197000 (36) | 0.0507 (36) | 9.47 (48)注4) |
| 15mg (N=9) | 24.08 (8.00-47.50) | 122 (103-148)注4) | 2370 (21) | 288000 (21) | 0.0502 (22)注5) | 9.43 (19)注4) |
図1)日本人2型糖尿病患者の血漿中チルゼパチド濃度(平均値+標準偏差)
日本人2型糖尿病患者11例に本剤5mgを週1回皮下投与したとき、薬物動態パラメータは表2のとおりであった3)。
| 投与量 | 本剤投与 | 例数 | tmax(h)注6) | t1/2(h)注7) | Cmax(ng/mL) | AUC(0-168)(ng・h/mL) | CL/F(L/h) | Vz/F(L) |
| 5mg | 1回目 | 11 | 48.00 (23.98-72.00) | − | 364 (20) | 48800 (16) | − | − |
| 8回目 | 11 | 48.00 (23.83-48.00) | 127 (112-144) | 838 (22) | 104000 (19) | 0.029 (21) | 5.27 (15) |
16.2 吸収
16.3 分布
16.3.1 蛋白結合
本剤は主に血漿アルブミンと強く結合(結合率:99.06%)する6)。
16.3.2 乳汁中への移行
授乳中の健康な女性11例に本剤5mgを単回皮下投与したとき、3例で乳汁中にチルゼパチドが検出され、乳汁中濃度範囲は4.6〜7.2ng/mLであった。[9.6参照]
16.4 代謝
本剤の代謝経路は、一般的なタンパク質の異化経路によるペプチド骨格の分解、C20脂肪酸部分のβ酸化及びアミド加水分解である。
16.5 排泄
本剤は代謝され主に尿中及び糞便中に排泄される。未変化体は尿中及び糞便中には認められなかった7)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
腎機能正常被験者(eGFR≧90mL/min/1.73m2)14例、軽度腎機能障害患者(eGFR:60〜89mL/min/1.73m2)8例、中等度腎機能障害患者(eGFR:30〜59mL/min/1.73m2)8例、重度腎機能障害患者(eGFR<30mL/min/1.73m2)7例及び末期腎疾患患者(3ヵ月以上血液透析を受けている)8例に本剤5mgを単回皮下投与した試験において、腎機能正常被験者に対する軽度、中等度、重度腎機能障害患者及び末期腎疾患患者の本剤のAUC(0-∞)の最小二乗幾何平均値の比[90%信頼区間]は、それぞれ1.05[0.86,1.27]、1.29[1.07,1.56]、1.03[0.84,1.27]及び1.16[0.96,1.40]であった。また、Cmaxの最小二乗幾何平均値の比[90%信頼区間]は、それぞれ1.04[0.84,1.30]、1.09[0.87,1.36]、1.23[0.97,1.56]及び1.02[0.82,1.27]であった8)(外国人データ)。
16.6.2 肝機能障害患者
肝機能正常被験者13例、軽度肝機能障害患者(Child-Pugh分類A)6例、中等度肝機能障害患者(Child-Pugh分類B)6例、重度肝機能障害患者(Child-Pugh分類C)7例に本剤5mgを単回皮下投与した試験において、肝機能正常被験者に対する軽度、中等度及び重度肝機能障害患者の本剤のAUC(0-∞)の最小二乗幾何平均値の比[90%信頼区間]は、それぞれ1.08[0.88,1.32]、0.96[0.79,1.17]及び0.85[0.70,1.04]であった。また、Cmaxの最小二乗幾何平均値の比[90%信頼区間]は、それぞれ0.92[0.73,1.16]、1.00[0.80,1.25]及び0.97[0.78,1.21]であった9)(外国人データ)。
16.6.3 高齢者
国内外の臨床試験19試験より得られた5802例(日本人1086例)を対象とした母集団薬物動態解析の結果、65歳未満と65歳以上の患者の薬物動態の間に大きな違いは認められないものと推定された10)。
16.7 薬物相互作用
本剤と経口避妊薬又はアセトアミノフェンを併用した薬物相互作用試験の結果を表3に示す11)12)(外国人データ)。[10.2、18.2.6参照]
| 併用薬 | 本剤投与量 | 本剤投与 | 例数 | 併用薬に対する影響 | ||
| Cmax比 [信頼区間] | AUC比 [信頼区間] | tmax差(hr) [信頼区間] | ||||
| 健康成人女性に本剤を単回投与 | ||||||
| 経口避妊薬注8) | ||||||
| ノルエルゲストロミン注9) | 5mg | 単回 | 25/25 | 0.45 [0.40,0.51] | 0.78 [0.71,0.84] | 4.50 [1.50,5.00] |
| エチニルエストラジオール | 24/24 | 0.41 [0.36,0.47] | 0.79注10) [0.73,0.85] | 4.23 [1.50,6.50] | ||
| 2型糖尿病患者に本剤を週1回反復投与 | ||||||
| アセトアミノフェン1g注11) [18.2.6参照] | 0.5mg | 1回目 | 9/11 | 1.10 [0.83,1.45] | 1.11 [0.88,1.39] | 0.00 [−1.00,1.00] |
| 4回目 | 9/11 | 1.15 [0.87,1.52] | 1.09 [0.87,1.37] | −0.17 [−1.00,1.00] | ||
| 5mg | 1回目 | 8/11 | 0.50 [0.37,0.66] | 0.75 [0.59,0.95] | 1.00 [0.00,2.25] | |
| 4回目 | 6/11 | 0.92 [0.67,1.26] | 1.05 [0.82,1.36] | 0.83 [−1.00,2.00] | ||
| 10mg注12) | 4回目 | 11/11 | 0.64 [0.49,0.83] | 1.04 [0.84,1.29] | 1.00 [0.00,2.00] | |
| 15mg注13) | 4回目 | 10/11 | 0.60 [0.46,0.79] | 1.07 [0.86,1.33] | 1.00 [1.00,2.00] | |
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 単独療法:プラセボ対照二重盲検比較試験(第III相国際共同試験)
食事・運動療法で血糖コントロールが不十分な2型糖尿病患者478例を対象に無作為割り付けを行い、二重盲検下で本剤5mg、10mg、15mg又はプラセボを週1回、40週間投与した(本剤5mg群:121例(日本人:23例)、本剤10mg群:121例(日本人:22例)、本剤15mg群:121例(日本人:23例)、プラセボ群:115例(日本人:21例))。本剤は、いずれの用量においても週1回2.5mgで投与を開始し、以後4週間ごとに2.5mgずつ増量した。
本剤5mg、10mg又は15mgの40週間投与により、主要評価項目であるHbA1cのベースラインから投与後40週までの変化量に関して、本剤のいずれの用量でもプラセボに対する優越性が検証された(p<0.001)13)。
本剤5mg、10mg又は15mgの40週間投与により、主要評価項目であるHbA1cのベースラインから投与後40週までの変化量に関して、本剤のいずれの用量でもプラセボに対する優越性が検証された(p<0.001)13)。
| HbA1c(%) | 本剤5mg | 本剤10mg | 本剤15mg | プラセボ |
| ベースライン注1) | 7.97±0.84 (121) | 7.88±0.77 (118) | 7.88±1.03 (116) | 8.08±0.80 (112) |
| 投与後40週までの変化量注2) | −1.87±0.09 (108) | −1.89±0.10 (105) | −2.07±0.10 (92) | 0.04±0.11 (70) |
| 群間差(本剤−プラセボ)注3) [95%信頼区間] | −1.91 [−2.18,−1.63] | −1.93 [−2.21,−1.65] | −2.11 [−2.39,−1.83] | − |
ベースラインから投与後40週までの体重の変化量(最小二乗平均±標準誤差)は、本剤5mg群で−7.0±0.52kg(ベースラインの平均±標準偏差:87.0±21.2kg)、本剤10mg群で−7.8±0.53kg(ベースラインの平均±標準偏差:85.7±18.9kg)、本剤15mg群で−9.5±0.54kg(ベースラインの平均±標準偏差:85.9±18.5kg)、プラセボ群で−0.7±0.57kg(ベースラインの平均±標準偏差:84.4±20.1kg)であった。
重症低血糖は報告されず、血糖値54mg/dL未満の低血糖は、プラセボ群で1/115例(0.9%)報告された。副作用発現頻度は、本剤5mg群で33.9%(41/121例)、本剤10mg群で46.3%(56/121例)、本剤15mg群で41.3%(50/121例)及びプラセボ群で23.5%(27/115例)であった。主な副作用は本剤5mg群では悪心10.7%(13/121例)、本剤10mg群では悪心12.4%(15/121例)及び下痢11.6%(14/121例)、本剤15mg群では悪心17.4%(21/121例)及び下痢10.7%(13/121例)であった。なお、本剤群で認められた主な副作用について、プラセボ群では悪心5.2%(6/115例)及び下痢5.2%(6/115例)であった。[11.1.1参照]
17.1.2 単独療法:実薬対照二重盲検比較試験(第III相国内試験)
食事・運動療法で血糖コントロールが不十分な日本人2型糖尿病患者636例を対象に無作為割り付けを行い、二重盲検下で本剤5mg、10mg、15mg又はデュラグルチド0.75mgを週1回、52週間投与した(本剤5mg群:159例、本剤10mg群:158例、本剤15mg群:160例、デュラグルチド群:159例)。本剤は、いずれの用量においても週1回2.5mgで投与を開始し、以後4週間ごとに2.5mgずつ増量した。
本剤5mg、10mg又は15mgの52週間投与により、主要評価項目であるHbA1cのベースラインから投与後52週までの変化量に関して、本剤のいずれの用量でもデュラグルチド0.75mgに対する優越性が検証された(p<0.001)14)。
本剤5mg、10mg又は15mgの52週間投与により、主要評価項目であるHbA1cのベースラインから投与後52週までの変化量に関して、本剤のいずれの用量でもデュラグルチド0.75mgに対する優越性が検証された(p<0.001)14)。
| HbA1c(%) | 本剤5mg | 本剤10mg | 本剤15mg | デュラグルチド0.75mg |
| ベースライン注4) | 8.17±0.88 (158) | 8.20±0.86 (156) | 8.20±0.89 (159) | 8.15±0.86 (159) |
| 投与後52週までの変化量注5) | −2.37±0.07 (142) | −2.55±0.07 (135) | −2.82±0.07 (134) | −1.29±0.07 (138) |
| 群間差(本剤−デュラグルチド)注6) [95%信頼区間] | −1.09 [−1.27,−0.90] | −1.27 [−1.45,−1.08] | −1.53 [−1.71,−1.35] | − |
ベースラインから投与後52週までの体重の変化量(最小二乗平均±標準誤差)は、本剤5mg群で−5.8±0.41kg(ベースラインの平均±標準偏差:78.6±16.4kg)、本剤10mg群で−8.5±0.42kg(ベースラインの平均±標準偏差:79.1±13.7kg)、本剤15mg群で−10.7±0.41kg(ベースラインの平均±標準偏差:78.9±14.3kg)、デュラグルチド群で−0.5±0.41kg(ベースラインの平均±標準偏差:76.5±13.2kg)であった。
重症低血糖は報告されず、血糖値54mg/dL未満の低血糖は、本剤15mg群で2/160例(1.3%)報告された。副作用発現頻度は、本剤5mg群52.8%(84/159例)、本剤10mg群で51.9%(82/158例)、本剤15mg群で60.6%(97/160例)及びデュラグルチド群で37.1%(59/159例)であった。主な副作用は本剤5mg群では便秘13.8%(22/159例)、食欲減退13.8%(22/159例)、悪心11.9%(19/159例)及び下痢10.1%(16/159例)、本剤10mg群では悪心19.6%(31/158例)、便秘16.5%(26/158例)及び食欲減退12.0%(19/158例)、本剤15mg群では食欲減退21.3%(34/160例)、悪心19.4%(31/160例)及び便秘11.9%(19/160例)であった。なお、本剤群で認められた主な副作用について、デュラグルチド群では便秘8.8%(14/159例)、悪心6.9%(11/159例)、下痢3.8%(6/159例)、食欲減退4.4%(7/159例)であった。[11.1.1参照]
また、収縮期血圧が90mmHg以下かつベースラインから20mmHg以上の低下が認められた被験者の割合は、本剤5mg群では1.9%(3/159例)、本剤10mg群では3.2%(5/158例)、本剤15mg群では5.0%(8/160例)であり、デュラグルチド0.75mg群では認められなかった。[8.13参照]
17.1.3 併用療法:基礎インスリンとの併用、プラセボ対照二重盲検比較試験(第III相国際共同試験)
基礎インスリンの単独療法又は基礎インスリンとメトホルミンとの併用療法で血糖コントロールが不十分な2型糖尿病患者475例を対象に無作為割り付けを行い、二重盲検下で本剤5mg、10mg、15mg又はプラセボを週1回、40週間追加投与した(本剤5mg群:116例(日本人:19例)、本剤10mg群:119例(日本人:21例)、本剤15mg群:120例(日本人:20例)、プラセボ群:120例(日本人:22例))。本剤は、いずれの用量においても週1回2.5mgで投与を開始し、以後4週間ごとに2.5mgずつ増量した。
本剤5mg、10mg又は15mgの40週間投与により、主要評価項目であるHbA1cのベースラインから投与後40週までの変化量に関して、本剤のいずれの用量でもプラセボに対する優越性が検証された(p<0.001)15)。
本剤5mg、10mg又は15mgの40週間投与により、主要評価項目であるHbA1cのベースラインから投与後40週までの変化量に関して、本剤のいずれの用量でもプラセボに対する優越性が検証された(p<0.001)15)。
| HbA1c(%) | 本剤5mg | 本剤10mg | 本剤15mg | プラセボ |
| ベースライン注7) | 8.29±0.88 (115) | 8.34±0.82 (113) | 8.22±0.85 (117) | 8.39±0.83 (118) |
| 投与後40週までの変化量注8) | −2.23±0.08 (105) | −2.59±0.08 (105) | −2.59±0.08 (97) | −0.93±0.08 (111) |
| 群間差(本剤−プラセボ)注9) [95%信頼区間] | −1.30 [−1.52,−1.07] | −1.66 [−1.88,−1.43] | −1.65 [−1.88,−1.43] | − |
ベースラインから投与後40週までの体重の変化量(最小二乗平均±標準誤差)は、本剤5mg群で−6.2±0.58kg、本剤10mg群で−8.2±0.58kg、本剤15mg群で−10.9±0.59kg、プラセボ群で1.7±0.57kgであった。
重症低血糖は、本剤10mg群で2/119例(1.7%)、本剤15mg群で1/120例(0.8%)報告された。血糖値54mg/dL未満の低血糖は、本剤5mg群で18/116例(15.5%)、本剤10mg群で23/119例(19.3%)、本剤15mg群で17/120例(14.2%)、プラセボ群で15/120例(12.5%)報告された。副作用発現頻度は、本剤5mg群で37.1%(43/116例)、本剤10mg群で38.7%(46/119例)、本剤15mg群で52.5%(63/120例)及びプラセボ群で14.2%(17/120例)であった。主な副作用は本剤5mg群では悪心12.9%(15/116例)及び下痢10.3%(12/116例)、本剤10mg群では悪心17.6%(21/119例)及び食欲減退12.6%(15/119例)、本剤15mg群では悪心17.5%(21/120例)、下痢16.7%(20/120例)、食欲減退14.2%(17/120例)及び嘔吐12.5%(15/120例)であった。なお、本剤群で認められた主な副作用について、プラセボ群では下痢4.2%(5/120例)及び食欲減退1.7%(2/120例)であり、悪心、及び嘔吐は認められなかった。[11.1.1参照]
17.1.4 非盲検長期(52週間)安全性試験(第III相国内試験)
経口血糖降下薬の単独療法で血糖コントロールが不十分な日本人2型糖尿病患者443例を対象に無作為割り付けを行い、本剤5mg、10mg又は15mgを週1回、経口血糖降下薬単剤(スルホニルウレア剤、ビグアナイド系薬剤、α-グルコシダーゼ阻害剤、チアゾリジン系薬剤、速効型インスリン分泌促進剤又はSGLT2阻害剤のいずれか)に52週間追加投与した(本剤5mg群:148例、本剤10mg群:147例、本剤15mg群:148例)。本剤は、いずれの用量においても週1回2.5mgで投与を開始し、以後4週間ごとに2.5mgずつ増量した。本試験の結果を下表に示す16)。
| HbA1c(%) | ベースライン注10) | 投与後52週までの変化量注11) | ||
| 本剤5mg | 本剤10mg | 本剤15mg | ||
| スルホニルウレア剤 | 8.69±1.05 (127) | −2.74±0.13 (42) | −2.95±0.13 (41) | −3.29±0.14 (38) |
| ビグアナイド系薬剤 | 8.44±0.96 (62) | −2.59±0.20 (19) | −3.02±0.19 (19) | −3.03±0.22 (16) |
| α-グルコシダーゼ阻害剤 | 8.55±1.31 (64) | −2.31±0.22 (18) | −2.94±0.22 (16) | −3.00±0.23 (14) |
| チアゾリジン系薬剤 | 8.37±1.08 (61) | −2.53±0.14 (19) | −2.93±0.14 (18) | −2.76±0.14 (20) |
| 速効型インスリン分泌促進剤 | 8.83±1.23 (62) | −2.66±0.24 (18) | −3.32±0.24 (20) | −3.23±0.24 (18) |
| SGLT2阻害剤 | 8.30±0.82 (63) | −2.31±0.19 (21) | −2.76±0.19 (20) | −2.66±0.19 (19) |
ベースラインから投与後52週までの体重の変化量(最小二乗平均)は、本剤5mg群、本剤10mg群及び本剤15mg群の順に、スルホニルウレア剤併用で−3.8、−6.5及び−8.5kg、ビグアナイド系薬剤併用で−4.4、−11.2及び−13.6kg、α-グルコシダーゼ阻害薬併用で−3.7、−8.9及び−8.0kg、チアゾリジン系薬剤併用で−2.1、−6.4及び−11.2kg、速効型インスリン分泌促進剤併用で−4.2、−6.5及び−9.7kg、SGLT-2阻害薬併用で−4.3、−6.4及び−11.6kgであった。
重症低血糖は報告されず、血糖値54mg/dL未満の低血糖は、本剤5mg群で1/148例(0.7%)(スルホニルウレア剤併用)、本剤10mg群で1/147例(0.7%)(速効型インスリン分泌促進剤併用)、本剤15mg群で3/148例(2.0%)(いずれもスルホニルウレア剤併用)報告された。副作用発現頻度は、本剤5mg群43.2%(64/148例)、本剤10mg群で52.4%(77/147例)及び本剤15mg群で63.5%(94/148例)であった。主な副作用は本剤5mg群では悪心8.8%(13/148例)、便秘7.4%(11/148例)及び食欲減退7.4%(11/148例)、本剤10mg群では悪心12.9%(19/147例)、便秘12.2%(18/147例)、下痢10.9%(16/147例)及び食欲減退10.2%(15/147例)、本剤15mg群では悪心25.7%(38/148例)、便秘14.2%(21/148例)、食欲減退12.2%(18/148例)、嘔吐9.5%(14/148例)及び下痢8.1%(12/148例)であった。[11.1.1参照]
18. 薬効薬理
18.1 作用機序
本剤はGIP受容体及びGLP-1受容体のアゴニストであり、両受容体に結合して活性化することで、グルコース濃度依存的にインスリン分泌を促進させる。本剤はC20脂肪酸側鎖を含む39個のアミノ酸からなるペプチドであり、内因性アルブミンと結合して消失半減期が延長することにより作用が持続する。
18.2 薬理作用
ヒトでの薬力学的作用の評価は、特記する場合を除き、すべて本剤15mgの週1回28週間(用量漸増期間を含む)皮下投与後の定常状態において行われた。
18.2.1 GIP受容体及びGLP-1受容体アゴニスト活性
本剤は、in vitro試験において、GIP受容体及びGLP-1受容体に結合して活性化し、いずれの受容体に対しても細胞内cAMPを増加させるアゴニスト活性を示した17)。
18.2.2 血糖降下作用
2型糖尿病患者に本剤を投与した結果、空腹時血糖及び食後血糖値はプラセボと比較して低下した18)(外国人データ)。
18.2.3 グルコース応答性インスリン分泌
マウス及びラット由来の単離膵島を用いたin vitro試験において、本剤はグルコース依存性インスリン分泌を促進した17)。ラットを用いたin vivo静脈内グルコース負荷試験において、本剤はグルコース依存性インスリン分泌を刺激した17)。
2型糖尿病患者に本剤を投与した結果、静脈内グルコース急速注入後のインスリンの第1相(グルコース投与直後から8分後)及び第2相(グルコース投与20分後から120分後)の分泌速度は、プラセボと比較して増加した18)(外国人データ)。
18.2.4 グルカゴン分泌
2型糖尿病患者に本剤を投与した結果、空腹時グルカゴン濃度及び食後のグルカゴン濃度のAUC(食後0〜4時間)はプラセボと比較して低下した18)(外国人データ)。
18.2.5 インスリン感受性
2型糖尿病患者に本剤を投与した結果、全身のインスリン感受性の指標であるM値のベースラインからの変化量はプラセボと比較して増加した18)(外国人データ)。
18.2.6 胃内容排出速度
2型糖尿病患者において、本剤5mg以上を週1回4週間反復投与した結果、アセトアミノフェンの血中濃度プロファイル(Cmax及びAUC)を指標とした胃内容排出速度の低下が認められた。胃内容排出速度の低下は初回投与後に最も顕著であり、4週間の反復投与で減弱した12)(外国人データ)。[9.1.6、16.7参照]
19. 有効成分に関する理化学的知見
19.1. チルゼパチド
| 一般的名称 | チルゼパチド |
|---|---|
| 一般的名称(欧名) | Tirzepatide |
| 分子式 | C225H348N48O68 |
| 分子量 | 4813.45 |
| 物理化学的性状 | 白色の粉末である。 |
| 理化学知見その他 | チルゼパチドは、ヒトグルコース依存性インスリン分泌刺激ポリペプチド(GIP)受容体及びヒトグルカゴン様ペプチド-1(GLP-1)受容体のアゴニストであり、2及び13番目のアミノ酸残基は2-methylAla、C末端はアミド化されたSerである。さらに、1,20-イコサン二酸が1個のGlu及び2個の8-アミノ-3,6-ジオキサオクタン酸で構成されるリンカーを介して20番目のLysに結合している。チルゼパチドは39個のアミノ酸残基からなる合成ペプチドである。 |
| KEGG DRUG |
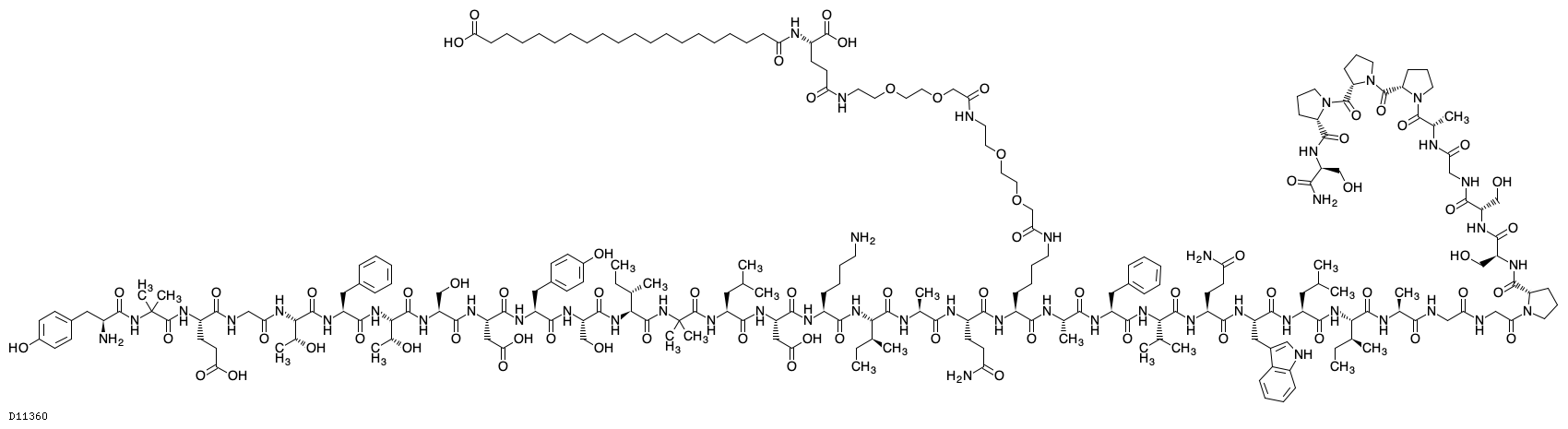
|
20. 取扱い上の注意
20.1 凍結を避け、2〜8℃で遮光保存すること。凍結した場合は、使用しないこと。
20.2 室温で保存する場合は、30℃を超えない場所で外箱から出さずに保存し、21日以内に使用すること。
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
<マンジャロ皮下注2.5mgアテオス>
0.5mL×2キット
<マンジャロ皮下注5mgアテオス>
0.5mL×2キット
<マンジャロ皮下注7.5mgアテオス>
0.5mL×2キット
<マンジャロ皮下注10mgアテオス>
0.5mL×2キット
<マンジャロ皮下注12.5mgアテオス>
0.5mL×2キット
<マンジャロ皮下注15mgアテオス>
0.5mL×2キット
23. 主要文献
- 社内資料:チルゼパチドの毒性試験(2022年9月26日承認、CTD2.6.6)
- 社内資料:日本人2型糖尿病患者の薬物動態(2022年9月26日承認、CTD2.7.2.2.2.1.2)
- Furihata K,et al., Diabetes Obes Metab., 24, 239-246, (2022) »PubMed
- 社内資料:投与部位の影響を評価した試験(2022年9月26日承認、CTD2.7.1.2.3)
- 社内資料:絶対的バイオアベイラビリティを評価した試験(2022年9月26日承認、CTD2.7.1.2.1)
- 社内資料:血漿蛋白結合(2022年9月26日承認、CTD2.7.2.2.1.1)
- 社内資料:マスバランス試験(2022年9月26日承認、CTD2.7.2.2.2.3.1)
- Urva S,et al., Clin Pharmacokinet., 60, 1049-1059, (2021) »PubMed
- Urva S,et al., Clin Pharmacokinet., 61, 1057-1067, (2022) »PubMed
- 社内資料:母集団薬物動態解析(2022年9月26日承認、CTD2.7.2.3〜2.7.2.3.7.8)
- 社内資料:経口避妊薬との薬物相互作用試験(GPGR試験)(2022年9月26日承認、CTD2.7.2.2.2.5.1)
- Urva S,et al., Diabetes Obes Metab., 22 (10), 1886-1891, (2020) »PubMed
- Rosenstock J,et al., Lancet., 398, 143-155, (2021) »PubMed
- Inagaki N,et al., Lancet Diabetes Endocrinol., 10 (9), 623-633, (2022) »PubMed
- Dahl D,et al., JAMA., 327 (6), 534-545, (2022) »PubMed
- Kadowaki T,et al., Lancet Diabetes Endocrinol., 10 (9), 634-644, (2022) »PubMed
- 社内資料:チルゼパチドの薬理試験(2022年9月26日承認、CTD2.6.2)
- Heise T,et al., Lancet Diabetes Endocrinol., 10, 418-429, (2022) »PubMed
24. 文献請求先及び問い合わせ先
文献請求先
日本イーライリリー株式会社
医薬情報問合せ窓口
〒651-0086
神戸市中央区磯上通5丁目1番28号
電話:0120-360-605(医療関係者向け)
田辺ファーマ株式会社
くすり相談センター
〒541-8505
大阪市中央区道修町3-2-10
電話:0120-753-280(医療関係者向け)
製品情報問い合わせ先
日本イーライリリー株式会社
医薬情報問合せ窓口
〒651-0086
神戸市中央区磯上通5丁目1番28号
電話:0120-360-605(医療関係者向け)
田辺ファーマ株式会社
くすり相談センター
〒541-8505
大阪市中央区道修町3-2-10
電話:0120-753-280(医療関係者向け)
26. 製造販売業者等
26.1 製造販売元
日本イーライリリー株式会社
神戸市中央区磯上通5丁目1番28号
26.2 販売元
田辺ファーマ株式会社
大阪市中央区道修町3-2-10