医薬品情報
| 総称名 | コセルゴ |
|---|---|
| 一般名 | セルメチニブ硫酸塩 |
| 欧文一般名 | Selumetinib Sulfate |
| 製剤名 | セルメチニブ硫酸塩 |
| 薬効分類名 | 神経線維腫症1型治療剤(MEK阻害剤) |
| 薬効分類番号 | 4299 |
| ATCコード | L01EE04 |
| KEGG DRUG |
D10024
セルメチニブ硫酸塩
|
| KEGG DGROUP |
DG03137
MEK阻害薬
|
| JAPIC | 添付文書(PDF) |
添付文書情報2025年9月 改訂(用法・用量変更)(第5版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| コセルゴカプセル10mg | Koselugo Capsules | アレクシオンファーマ | 4299004M1025 | 12622.8円/カプセル | 劇薬, 処方箋医薬品 |
| コセルゴカプセル25mg | Koselugo Capsules | アレクシオンファーマ | 4299004M2021 | 30257.8円/カプセル | 劇薬, 処方箋医薬品 |
1. 警告
本剤は、緊急時に十分対応できる医療施設において、本剤についての十分な知識と神経線維腫症1型の治療の十分な知識・経験を持つ医師のもとで、本剤の投与が適切と判断される症例についてのみ投与すること。
2. 禁忌
次の患者には投与しないこと
4. 効能または効果
5. 効能または効果に関連する注意
6. 用法及び用量
通常、セルメチニブとして1回25mg/m2(体表面積)を1日2回経口投与するが、患者の状態により適宜減量する。ただし、1回量は50mgを上限とする。
7. 用法及び用量に関連する注意
7.2 体表面積から換算した本剤の投与量は以下の表のとおりとする。
| 体表面積(m2) | 投与量 |
| 0.55-0.69 | 朝20mg/夜10mg |
| 0.70-0.89 | 1回20mg 1日2回 |
| 0.90-1.09 | 1回25mg 1日2回 |
| 1.10-1.29 | 1回30mg 1日2回 |
| 1.30-1.49 | 1回35mg 1日2回 |
| 1.50-1.69 | 1回40mg 1日2回 |
| 1.70-1.89 | 1回45mg 1日2回 |
| ≧1.90 | 1回50mg 1日2回 |
7.3 本剤投与により副作用が発現した場合には、以下の基準を参考に、本剤を休薬、減量又は中止すること。2段階減量後に忍容性が認められない場合、投与を中止すること。
| 体表面積(m2) | 1段階減量(1回用量) | 2段階減量(1回用量) | ||
| 朝 | 夜 | 朝 | 夜 | |
| 0.55-0.69 | 10mg | 10mg | 10mg/日 | |
| 0.70-0.89 | 20mg | 10mg | 10mg | 10mg |
| 0.90-1.09 | 25mg | 10mg | 10mg | 10mg |
| 1.10-1.29 | 25mg | 20mg | 20mg | 10mg |
| 1.30-1.49 | 25mg | 25mg | 25mg | 10mg |
| 1.50-1.69 | 30mg | 30mg | 25mg | 20mg |
| 1.70-1.89 | 35mg | 30mg | 25mg | 20mg |
| ≧1.90 | 35mg | 35mg | 25mg | 25mg |
| 副作用 | 程度注) | 処置 |
| 左室駆出率(LVEF)低下 | 投与前から10%以上低下かつ正常下限値以下で無症候性 | 回復するまで休薬し、再開する場合、用量を1段階減量して投与する。 |
| 症候性又はGrade3以上 | 投与を中止する。 | |
| 眼障害 | 網膜色素上皮剥離又は中心性漿液性網膜症 | 回復するまで休薬し、再開する場合、用量を1段階減量して投与する。 |
| 網膜静脈閉塞 | 投与を中止する。 | |
| 筋障害 | Grade1又は忍容可能なGrade2のCK上昇又は筋症状 | 患者の状態に注意しながら投与を継続する。 |
| 忍容不能なGrade2又はGrade3のCK上昇又は筋症状 | Grade1以下に回復するまで休薬し、再開する場合、用量を1段階減量して投与する。 | |
| Grade4のCK上昇 | Grade1以下に回復するまで休薬し、再開する場合、用量を1段階減量して投与する。また、投与中止を検討する。 | |
| 横紋筋融解症 | 投与を中止する。 | |
| 下痢 | Grade1又は忍容可能なGrade2 | 患者の状態に注意しながら投与を継続する。 |
| 忍容不能なGrade2又はGrade3 | Grade1以下に回復するまで休薬し、再開する場合、用量を1段階減量して投与する。 | |
| Grade4 | 投与を中止する。 | |
| 上記以外の副作用 | Grade1又は忍容可能なGrade2 | 患者の状態に注意しながら投与を継続する。 |
| 忍容不能なGrade2又はGrade3 | Grade1以下に回復するまで休薬し、再開する場合、用量を1段階減量して投与する。 | |
| Grade4 | Grade1以下に回復するまで休薬し、再開する場合、用量を1段階減量して投与する。また、投与中止を検討する。 |
7.5 強い又は中程度のCYP3A阻害剤若しくはフルコナゾールとの併用は可能な限り避けること。やむを得ず併用する場合には、以下の表に従い、1回20mg/m2の1日2回投与とし、併用中に副作用が発現した場合には、1回15mg/m2の1日2回投与に減量すること。[10.2、16.7.1、16.7.2、16.7.4参照]
| 体表面積(m2) | 20mg/m2(1回用量) | 15mg/m2(1回用量) | ||
| 朝 | 夜 | 朝 | 夜 | |
| 0.55-0.69 | 10mg | 10mg | 10mg/日 | |
| 0.70-0.89 | 20mg | 10mg | 10mg | 10mg |
| 0.90-1.09 | 20mg | 20mg | 20mg | 10mg |
| 1.10-1.29 | 25mg | 25mg | 25mg | 10mg |
| 1.30-1.49 | 30mg | 25mg | 25mg | 20mg |
| 1.50-1.69 | 35mg | 30mg | 25mg | 25mg |
| 1.70-1.89 | 35mg | 35mg | 30mg | 25mg |
| ≧1.90 | 40mg | 40mg | 30mg | 30mg |
7.6 10mgカプセルと25mgカプセルの生物学的同等性は示されていないため、1回50mgを投与する際には10mgカプセルを使用しないこと。
7.7 本剤とセルメチニブ顆粒の生物学的同等性は示されていない。本剤とセルメチニブ顆粒の切替えを行う場合は、患者の状態をより慎重に観察すること。
8. 重要な基本的注意
8.2 眼障害があらわれることがあるので、本剤投与中は定期的に眼の異常の有無を確認すること。また、眼の異常が認められた場合には、速やかに医療機関を受診するよう患者を指導すること。[11.1.2参照]
8.3 肝機能障害があらわれることがあるので、本剤投与中は定期的に肝機能検査を行うこと。[11.1.4参照]
8.4 横紋筋融解症、ミオパチーがあらわれることがあるので、本剤投与中は定期的にCK、クレアチニン等の検査を行い、筋肉痛、脱力感、CK上昇、血中及び尿中ミオグロビン上昇等に十分注意すること。[11.1.5参照]
8.5 貧血、ヘモグロビン減少、好中球減少、リンパ球減少、血小板減少があらわれることがあるので、本剤投与中は定期的に血液検査(血球数算定、白血球分画等)を行うこと。[11.1.6参照]
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.3 肝機能障害患者
9.3.1 重度の肝機能障害患者(Child-Pugh分類C)
9.3.2 中等度の肝機能障害患者(Child-Pugh分類B)
9.4 生殖能を有する者
9.4.1 妊娠可能な女性に対しては、本剤投与中及び投与終了後1ヵ月間は適切な避妊を行うよう指導すること。[9.5参照]
9.4.2 パートナーが妊娠する可能性がある男性に対しては、本剤投与中及び投与終了後1週間は適切な避妊を行うよう指導すること。
9.5 妊婦
9.6 授乳婦
授乳しないことが望ましい。本剤又は本剤の代謝物がヒトの母乳中に移行するかどうかは不明であるが、動物試験(マウス)で授乳中の母動物へ本剤を投与した際、本剤及び本剤の代謝物が乳汁中に排泄されることが認められている。
9.7 小児等
10. 相互作用
相互作用序文
本剤は、主にCYP3Aにより代謝され、CYP2C19も関与する。[16.4参照]
薬物代謝酵素用語
CYP3A
薬物代謝酵素用語
CYP2C19
10.2 併用注意
| 強い又は中程度のCYP3A阻害剤 クラリスロマイシン エリスロマイシン イトラコナゾール等 グレープフルーツジュース [7.5、16.7.1、16.7.4参照] | 本剤の副作用が増強されるおそれがあるため、これらの薬剤との併用は可能な限り避けること。 やむを得ず併用する場合には、減量するとともに、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | これらの薬剤等がCYP3Aを阻害することにより、本剤の代謝が阻害され、本剤の血中濃度が上昇する可能性がある。 |
| フルコナゾール [7.5、16.7.2参照] | 本剤の副作用が増強されるおそれがあるため、これらの薬剤との併用は可能な限り避けること。 やむを得ず併用する場合には、減量するとともに、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | CYP2C19及びCYP3Aを阻害することにより、本剤の代謝が阻害され、本剤の血中濃度が上昇する可能性がある。 |
| 強い又は中程度のCYP3A誘導剤 フェニトイン リファンピシン カルバマゼピン等 [16.7.3、16.7.4参照] | 本剤の効果が減弱するおそれがあるため、これらの薬剤との併用は可能な限り避けること。 | これらの薬剤等がCYP3Aを誘導することにより、本剤の代謝が促進され、本剤の血中濃度が低下する可能性がある。 |
| セイヨウオトギリソウ(St.John's Wort、セント・ジョーンズ・ワート)含有食品 | 本剤の効果が減弱するおそれがあるため、摂取しないよう注意すること。 | これらの薬剤等がCYP3Aを誘導することにより、本剤の代謝が促進され、本剤の血中濃度が低下する可能性がある。 |
| ビタミンE含有製剤(サプリメント等) | ビタミンEの摂取を控えるよう指導すること。 | 添加剤であるコハク酸トコフェロールポリエチレングリコールとして、本剤10mgには32mg、本剤25mgには36mgのビタミンEが含まれる。ビタミンEの高用量摂取により、出血のリスクを増強させる可能性がある。 |
| 抗凝固剤 抗血小板剤 ワルファリン アスピリン等 | プロトロンビン時間国際標準比(INR)値等の血液凝固能の検査、臨床症状の観察を頻回に行い、これらの薬剤の用量を調節すること。 | 添加剤であるコハク酸トコフェロールポリエチレングリコールとして、本剤10mgには32mg、本剤25mgには36mgのビタミンEが含まれる。ビタミンEの高用量摂取により、出血のリスクを増強させる可能性がある。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 心機能障害
11.1.2 眼障害
網膜色素上皮剥離(頻度不明)、中心性漿液性網膜症(頻度不明)、網膜静脈閉塞(頻度不明)等の眼障害があらわれることがある。[8.2参照]
11.1.3 消化管障害
下痢(31.1%)、嘔吐(25.1%)、悪心(21.3%)等の消化管障害があらわれることがある。
11.1.4 肝機能障害
AST(17.4%)、ALT(14.0%)、ビリルビン(0.4%)等の上昇を伴う肝機能障害があらわれることがある。[8.3参照]
11.1.5 横紋筋融解症(頻度不明)[8.4参照]
11.1.6 貧血及び血球減少
貧血(13.6%)、好中球減少(7.2%)、リンパ球減少(3.8%)、血小板減少(2.6%)等があらわれることがある。[8.5参照]
11.1.7 間質性肺疾患(頻度不明)
副作用発現頻度は本剤及びセルメチニブ顆粒の臨床試験成績に基づく。
11.2 その他の副作用
| 20%以上 | 10%〜20%未満 | 1%〜10%未満 | |
| 眼 | − | − | 霧視 |
| 呼吸器 | − | − | 呼吸困難 |
| 消化器 | 口内炎 | − | 便秘、口内乾燥 |
| 皮膚 | ざ瘡様皮膚炎(46.4%)、爪囲炎、発疹、皮膚乾燥、脱毛・毛髪変色 | − | − |
| その他 | 血中CK増加(40.9%) | 疲労・無力症、末梢性浮腫 | 低アルブミン血症、顔面浮腫、発熱、血中クレアチニン増加、高血圧 |
14. 適用上の注意
14.1 薬剤調製時の注意
吸湿により添加剤が加水分解され本剤の品質に影響を及ぼす可能性があるため、分包せずボトルのまま交付すること。
14.2 薬剤交付時の注意
15. その他の注意
15.2 非臨床試験に基づく情報
マウスを用いた反復投与毒性試験において、臨床曝露量の約22倍で盲腸及び結腸の穿孔が認められ、回復性は確認されていない。また、ラットを用いた反復投与毒性試験において、臨床曝露量の約9倍で骨端軟骨異形成が認められ、回復性は確認されていない。
16. 薬物動態
16.1 血中濃度
16.1.1 国内第I相試験(D1346C00013試験)
3歳以上18歳以下の叢状神経線維腫を有する日本人神経線維腫症1型患者12例に本剤25mg/m2を1日2回空腹時に反復経口投与したとき、投与1日目及び29日目のセルメチニブ及び活性代謝物であるN-脱メチル体の血漿中濃度推移及び薬物動態パラメータは以下のとおりであった1)。
3歳以上18歳以下の叢状神経線維腫を有する日本人神経線維腫症1型患者に本剤25mg/m2を1日2回空腹時に反復経口投与したときの血漿中濃度推移(算術平均値+標準偏差)
| 測定対象 | 測定日 | 例数 | Cmax(ng/mL) | Tmaxa)(h) | AUC0-6h(ng・h/mL) | AUC0-12h(ng・h/mL) |
| セルメチニブ | 投与1日目 | 12 | 783.1(52.70) | 1.49[0.50,3.05] | 1926(41.64) | 2523(24.23)b) |
| 投与29日目 | 11 | 869.4(53.53) | 1.47[0.42,2.87] | 2396(40.32) | − | |
| N-脱メチル体 | 投与1日目 | 12 | 55.47(49.13) | 1.52[1.37,3.05] | 140.8(38.86) | 188.7(24.32)c) |
| 投与29日目 | 11 | 40.90(53.10) | 1.47[0.42,2.87] | 123.6(37.34) | − |
16.1.2 海外第I相試験(D1532C00057試験第I相パート)
3歳以上18歳以下の叢状神経線維腫を有する外国人神経線維腫症1型患者13例に本剤20又は25mg/m2を空腹時に単回経口投与したとき注)のセルメチニブ及び活性代謝物であるN-脱メチル体の薬物動態パラメータは以下のとおりであった。
| 測定対象 | 投与量 | 例数 | Cmax(ng/mL) | Tmaxa)(h) | AUC0-12h(ng・h/mL) | T1/2b)(h) |
| セルメチニブ | 20mg/m2 | 9 | 754.7(47.53) | 1.08[0.98,3.00] | 1898(18.83) | 9.41±4.59 |
| 25mg/m2 | 4 | 928.4(18.17) | 1.04[1.00,2.00] | 2199(14.83) | 6.16±0.88 | |
| N-脱メチル体 | 20mg/m2 | 9 | 58.05(70.87) | 1.08[0.98,3.00] | 163.3(28.84) | 9.44±10.24 |
| 25mg/m2 | 4 | 56.02(35.27) | 1.04[1.00,2.00] | 150.9(24.33) | 4.47±2.49 |
16.1.3 国際共同第III相試験(D134BC00001試験/KOMET)
18歳以上の日本人及び外国人の叢状神経線維腫を有する神経線維腫症1型患者64例(うち、日本人6例)に本剤25mg/m2を1日2回空腹時に反復経口投与したとき、投与8日目のセルメチニブ及び活性代謝物であるN-脱メチル体の薬物動態パラメータは以下のとおりであった2)。
| 測定対象 | 患者集団 | 例数 | Cmax(ng/mL) | Tmaxa)(h) | AUC0-6h(ng・h/mL) | AUC0-12h(ng・h/mL) |
| セルメチニブ | 全体 | 64 | 788.8(46.88) | 1.50[0.50,5.97] | 2224(41.56)b) | 2986(42.65)b) |
| 日本人 | 6 | 915.4(37.15) | 1.48[1.38,3.00] | 2614(24.82) | 3547(24.04) | |
| N-脱メチル体 | 全体 | 64 | 39.47(54.68) | 1.50[1.38,5.97] | 114.3(49.48)b) | 159.3(49.15)c) |
| 日本人 | 6 | 46.95(45.60) | 1.48[1.38,3.00] | 141.7(39.43) | 197.5(34.34) |
16.2 吸収
16.2.1 バイオアベイラビリティ
健康成人12例に本剤75mgを単回経口投与したとき注)、絶対バイオアベイラビリティは62%であった3)(外国人データ)。
16.2.2 食事の影響
健康成人34例に本剤75mgを高脂肪食の摂取後に単回経口投与したとき注)、絶食下投与と比較して、Cmaxは50%低下し、AUCinfは16%低下し、Tmaxは約1.5時間延長した4)(外国人データ)。
健康成人24例に本剤50mgを低脂肪食の摂取後に単回経口投与したとき注)、絶食下投与と比較して、Cmaxは60%低下し、AUCinfは38%低下し、Tmaxは約0.9時間延長した5)(外国人データ)。
12歳以上18歳未満の叢状神経線維腫を有する神経線維腫症1型患者25例に本剤25mg/m2を1日2回8日間低脂肪食の摂取後に反復経口投与したとき、空腹時投与と比較して、Cmaxは24%低下し、AUC0-12hは8%低下し、Tmaxは約1時間延長した6)(外国人データ)。
16.3 分布
In vitro試験において、セルメチニブのヒト血漿蛋白結合率は98.4%であった。セルメチニブは主にヒト血清アルブミンに対して結合し(96.1%)、α1-酸性糖蛋白への結合率は27.2%であった7)。
16.4 代謝
セルメチニブは主にCYP3Aにより代謝され、CYP2C19も関与する(代謝における推定寄与率:25%及び15%8))。また、セルメチニブのグルクロン酸抱合にはUGT1A1及びUGT1A3が寄与することが示唆された。[10.参照]
健康成人6例にセルメチニブの14C標識体75mgを単回経口投与したとき注)、ヒト血漿中の主なセルメチニブ関連成分は、未変化体のセルメチニブ(投与放射能の約40%)、イミダゾインダゾール体のグルクロン酸抱合体(22%)であった。その他、セルメチニブのグルクロン酸抱合体(7%)、N-脱メチルカルボン酸体(3.6%)、活性代謝物であるN-脱メチル体(2.9%)等が認められた9)(外国人データ)。
健康成人6例にセルメチニブの14C標識体75mgを単回経口投与したとき注)、ヒト血漿中の主なセルメチニブ関連成分は、未変化体のセルメチニブ(投与放射能の約40%)、イミダゾインダゾール体のグルクロン酸抱合体(22%)であった。その他、セルメチニブのグルクロン酸抱合体(7%)、N-脱メチルカルボン酸体(3.6%)、活性代謝物であるN-脱メチル体(2.9%)等が認められた9)(外国人データ)。
16.5 排泄
健康成人6例にセルメチニブの14C標識体75mgを単回経口投与したとき注)、投与後9日間で投与量の59%の放射能標識体が糞中から回収され(未変化体は投与量の19%)、33%が尿中から回収された(未変化体は投与量の1%未満)9)(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害
腎機能が正常な成人被験者11例及び末期腎不全の成人被験者12例に本剤50mgを単回経口投与したとき注)、腎機能が正常な被験者に比べて末期腎不全の被験者では、Cmaxは16%低く、AUCinfは28%低く、非結合形のCmaxは13%高く、非結合形のAUCinfは3%低かった。末期腎不全の被験者におけるN-脱メチル体のAUCinfはセルメチニブの約20%であり、腎機能が正常な被験者よりやや高かった10)(外国人データ)。
16.6.2 肝機能障害
肝機能が正常な成人被験者(8例)及び軽度の肝機能障害を有する成人被験者(Child-Pugh分類A、8例)に本剤50mgを、中等度の肝機能障害を有する成人被験者(Child-Pugh分類B、8例)に本剤50mg又は25mgを、並びに重度の肝機能障害を有する成人被験者(Child-Pugh分類C、8例)に本剤20mgを単回経口投与したとき注)、肝機能が正常な被験者に比べて軽度の肝機能障害を有する被験者では用量補正AUCinf及び用量補正非結合形AUCinfはそれぞれ86%及び69%であったが、中等度の肝機能障害を有する被験者ではそれぞれ159%及び141%、重度の肝機能障害を有する被験者ではそれぞれ157%及び317%と高かった10)(外国人データ)。[2.3、7.4、9.3.1、9.3.2参照]
16.7 薬物相互作用
16.7.1 イトラコナゾール
16.7.2 フルコナゾール
16.7.3 リファンピシン
16.7.4 エリスロマイシン、ジルチアゼム、エファビレンツ
注)本剤の承認用法及び用量は「通常、セルメチニブとして1回25mg/m2(体表面積)を1日2回経口投与するが、患者の状態により適宜減量する。ただし、1回量は50mgを上限とする。」である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 海外第II相試験(D1532C00057試験第II相パート層1)
3歳以上18歳以下の疼痛や外観上の変形、運動機能障害、気道機能障害等の臨床症状を有し生命維持に必要な構造を巻き込んでいる等により重大な合併症のリスクを伴うことなく完全に切除できない叢状神経線維腫(Plexiform neurofibroma)を有する神経線維腫症1型患者(悪性末梢神経鞘腫瘍を合併する患者は除外)50例を対象に、本剤1回25mg/m2(体表面積1.9m2以上の場合は50mg)を1日2回空腹時に経口投与する非盲検試験を実施した12)。主要評価項目であるResponse Evaluation in Neurofibromatosis and Schwannomatosis(REiNS)基準13)に基づく奏効率(例数)[95%信頼区間]は66.0%(33/50例)[51.2,78.8]であった注)。なお、最良総合効果は確定部分奏効33例、未確定部分奏効4例、及び安定11例であった。総投与期間の中央値は2.2年であった。(データカットオフ日:2018年6月29日)[5.1、7.1、9.7参照]
本剤が投与された50例中49例(98.0%)に有害事象が認められ、主な有害事象は、嘔吐43例(86.0%)、血中CK増加39例(78.0%)、下痢37例(74.0%)及び悪心36例(72.0%)であった。(データカットオフ日:2021年3月31日)
本剤が投与された50例中49例(98.0%)に有害事象が認められ、主な有害事象は、嘔吐43例(86.0%)、血中CK増加39例(78.0%)、下痢37例(74.0%)及び悪心36例(72.0%)であった。(データカットオフ日:2021年3月31日)
17.1.2 国内第I相試験(D1346C00013試験)
3歳以上18歳以下の疼痛や外観上の変形、運動機能障害等の臨床症状を有し生命維持に必要な構造を巻き込んでいる等により重大な合併症のリスクを伴うことなく完全に切除できない叢状神経線維腫(Plexiform neurofibroma)を有する神経線維腫症1型患者(悪性末梢神経鞘腫瘍を合併する患者は除外)12例を対象に、本剤1回25mg/m2(体表面積1.9m2以上の場合は50mg)を1日2回空腹時に経口投与する非盲検単群試験を実施した1)。REiNS基準に基づく奏効率(例数)[95%信頼区間]は33.3%(4/12例)[9.9,65.1]であり、最良総合効果は確定部分奏効4例(33.3%)、未確定部分奏効2例(16.7%)、安定4例(33.3%)、病勢進行2例(16.7%)であった注)。[5.1、7.1、9.7参照]
本剤が投与された12例全例に有害事象が認められ、主な有害事象は、湿疹7例(58.3%)、ざ瘡様皮膚炎6例(50.0%)、下痢及び爪囲炎各5例(41.7%)、嘔吐、皮膚乾燥及び口内炎各4例(33.3%)であった。(12カ月間投与データ)
本剤が投与された12例全例に有害事象が認められ、主な有害事象は、湿疹7例(58.3%)、ざ瘡様皮膚炎6例(50.0%)、下痢及び爪囲炎各5例(41.7%)、嘔吐、皮膚乾燥及び口内炎各4例(33.3%)であった。(12カ月間投与データ)
17.1.3 国際共同第III相試験(D134BC00001試験/KOMET)
18歳以上の疼痛や外観上の変形、運動機能障害、気道機能障害等の臨床症状を有し生命維持に必要な構造を巻き込んでいる等により重大な合併症のリスクを伴うことなく完全に切除できない叢状神経線維腫(Plexiform neurofibroma)を有する神経線維腫症1型患者(悪性末梢神経鞘腫瘍を合併する患者は除外)145例(日本人15例を含む)を対象に、本剤1回25mg/m2(体表面積1.9m2以上の場合は50mg)又はプラセボを1日2回空腹時に経口投与するプラセボ対照無作為化二重盲検比較試験を実施した2)。本試験ではサイクル12(1サイクル28日間)までを二重盲検期間とし、プラセボ群の患者はサイクル12終了後に本剤に切り替えた。主要評価項目であるREiNS基準に基づくサイクル16までの奏効率(例数)[95.3%信頼区間]は、本剤群で19.7%(14/71例)[11.1,31.0]、プラセボ群で5.4%(4/74例)[1.5,13.4]で、その群間差[95.3%信頼区間]は14.3%[3.7,26.0]であり、本剤群の奏効率はプラセボ群と比較して統計学的に有意に高かった(p=0.0112、有意水準両側0.047)。なお、サイクル16までの最良総合効果は本剤群で確定部分奏効14例(19.7%)、安定50例(70.4%)(未確定部分奏効5例[7.0%]を含む)、病勢進行1例(1.4%)、プラセボ群で確定部分奏効4例(5.4%)、安定63例(85.1%)(未確定部分奏効8例[10.8%]を含む)、病勢進行5例(6.8%)であった注)。二重盲検期間の総投与期間の中央値は本剤群336.0日、プラセボ群335.0日であった。[5.1参照]
二重盲検期間では本剤群の71例全例(100%)、プラセボ群の74例中68例(91.9%)に有害事象が認められ、主な有害事象は、本剤群でざ瘡様皮膚炎42例(59.2%)、血中CK増加32例(45.1%)、下痢30例(42.3%)、悪心及び嘔吐各18例(25.4%)であり、プラセボ群でCOVID-19 15例(20.3%)であった。
本剤が投与された137例中133例(97.1%)に有害事象が認められ、主な有害事象はざ瘡様皮膚炎64例(46.7%)、血中CK増加51例(37.2%)及び下痢41例(29.9%)であった。(データカットオフ日:2024年8月5日)
二重盲検期間では本剤群の71例全例(100%)、プラセボ群の74例中68例(91.9%)に有害事象が認められ、主な有害事象は、本剤群でざ瘡様皮膚炎42例(59.2%)、血中CK増加32例(45.1%)、下痢30例(42.3%)、悪心及び嘔吐各18例(25.4%)であり、プラセボ群でCOVID-19 15例(20.3%)であった。
本剤が投与された137例中133例(97.1%)に有害事象が認められ、主な有害事象はざ瘡様皮膚炎64例(46.7%)、血中CK増加51例(37.2%)及び下痢41例(29.9%)であった。(データカットオフ日:2024年8月5日)
注)完全奏効:標的病変の消失、部分奏効:標的叢状神経線維腫腫瘍容積がベースラインから20%以上減少、安定:ベースラインからの腫瘍容積の変化が部分奏効及び病勢進行の基準に合致しない、病勢進行:標的叢状神経線維腫腫瘍容積がベースライン又は最良効果判定時から20%以上増加。初回奏効後3カ月以降に実施した再評価で奏効を確定した。奏効率は、海外第II相試験及び国内第I相試験では完全奏効又は確定部分奏効が認められた患者の割合、国際共同第III相試験では確定完全奏効又は確定部分奏効が認められた患者の割合とした。
18. 薬効薬理
18.1 作用機序
セルメチニブは、MEK1/2を阻害することにより、MEKの基質であるERKのリン酸化を阻害し、RASにより調節されるRAF/MEK/ERK経路のシグナル伝達を抑制することで、NF1における神経線維腫の増殖を抑制すると考えられる14)。
18.2 神経線維腫の増殖抑制作用
シュワン細胞特異的にNF1遺伝子を欠失させた遺伝子改変マウス神経線維腫モデルにおいて、セルメチニブは神経線維腫組織内におけるERKのリン酸化を阻害し、神経線維腫の増殖を抑制した15)。
19. 有効成分に関する理化学的知見
19.1. セルメチニブ硫酸塩
| 一般的名称 | セルメチニブ硫酸塩 |
|---|---|
| 一般的名称(欧名) | Selumetinib Sulfate |
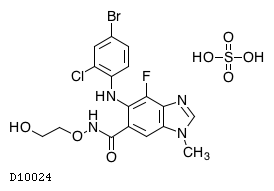
| 化学名 | 5-[(4-Bromo-2-chlorophenyl)amino]-4-fluoro-N-(2-hydroxyethoxy)-1-methyl-1H-benzimidazole-6-carboxamide monosulfate |
| 分子式 | C17H15BrClFN4O3・H2SO4 |
| 分子量 | 555.76 |
| 物理化学的性状 | 白色〜黄色の粉末である。メタノールに溶けにくく、エタノールに極めて溶けにくく、水にほとんど溶けない。 |
| KEGG DRUG |
|
20. 取扱い上の注意
21. 承認条件
21.1 医薬品リスク管理計画を策定の上、適切に実施すること。
<小児>
21.2 国内での治験症例が極めて限られていることから、製造販売後、一定数の症例に係るデータが集積されるまでの間は、全症例を対象に使用成績調査を実施することにより、本剤の使用患者の背景情報を把握するとともに、本剤の安全性及び有効性に関するデータを早期に収集し、本剤の適正使用に必要な措置を講じること。
22. 包装
<コセルゴカプセル10mg>
28カプセル[ボトル、バラ、乾燥剤入り]
<コセルゴカプセル25mg>
28カプセル[ボトル、バラ、乾燥剤入り]
23. 主要文献
- 社内資料:セルメチニブの小児期の神経線維腫症1型患者を対象とした国内第I相試験(D1346C00013)(2022年9月26日承認、CTD 2.7.6.2.1)
- 社内資料:セルメチニブの成人の神経線維腫症1型患者を対象とした国際共同第III相試験(D134BC00001/KOMET)(2025年8月25日承認、CTD 2.7.6.1)
- 社内資料:絶対バイオアベイラビリティ(D1532C00080)(2022年9月26日承認、CTD 2.7.6.2.17)
- Tomkinson H,et al., Clin Ther., 39 (11), 2260-2275.e1., (2017) »PubMed
- 社内資料:食事の影響(参考)(D1532C00089)(2022年9月26日承認、CTD 2.7.6.2.23)
- 社内資料:食事の影響(参考)(D1346C00015)(2025年9月19日承認、CTD 2.7.6.2)
- 社内資料:血漿蛋白結合(in vitro試験)(2022年9月26日承認、CTD 2.6.4.4.1)
- Cohen-Rabbie S,et al., J Clin Pharmacol., 61 (11), 1493-1504, (2021) »PubMed
- Dymond AW,et al., Clin Ther., 38 (11), 2447-2458, (2016) »PubMed
- Dymond AW,et al., J Clin Pharmacol., 57 (5), 592-605, (2017) »PubMed
- Dymond AW,et al., Eur J Clin Pharmacol., 73 (2), 175-184, (2017) »PubMed
- 社内資料:セルメチニブの小児期の神経線維腫症1型患者を対象とした海外第II相試験(D1532C00057試験第II相パート層1)(2022年9月26日承認、CTD 2.7.6.2.2)
- Dombi E,et al., Neurology., 81 (Suppl 1), S33-S40, (2013)
- Caunt CJ,et al., Nat Rev Cancer., 15 (10), 577-592, (2015) »PubMed
- Dombi E,et al., N Engl J Med., 375 (26), 2550-2260, (2016) »PubMed
24. 文献請求先及び問い合わせ先
文献請求先
アレクシオンファーマ合同会社
メディカル インフォメーション センター
〒108-0023
東京都港区芝浦三丁目1番1号 田町ステーションタワーN
電話:0120-577-657
製品情報問い合わせ先
アレクシオンファーマ合同会社
メディカル インフォメーション センター
〒108-0023
東京都港区芝浦三丁目1番1号 田町ステーションタワーN
電話:0120-577-657
26. 製造販売業者等
26.1 製造販売元
アレクシオンファーマ合同会社
〒108-0023
東京都港区芝浦三丁目1番1号 田町ステーションタワーN