医薬品情報
| 総称名 | ドプテレット |
|---|---|
| 一般名 | アバトロンボパグマレイン酸塩 |
| 欧文一般名 | Avatrombopag Maleate |
| 製剤名 | アバトロンボパグマレイン酸塩錠 |
| 薬効分類名 | トロンボポエチン受容体作動薬 |
| 薬効分類番号 | 3399 |
| ATCコード | B02BX08 |
| KEGG DRUG |
D10307
アバトロンボパグマレイン酸塩
|
| JAPIC | 添付文書(PDF) |
添付文書情報2026年4月 改訂(第7版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ドプテレット錠20mg | Doptelet tablets | Swedish Orphan Biovitrum Japan | 3399012F1021 | 7106.6円/錠 | 処方箋医薬品注) |
2. 禁忌
次の患者には投与しないこと
本剤の成分に対し過敏症の既往歴のある患者
4. 効能または効果
5. 効能または効果に関連する注意
<待機的な観血的手技を予定している慢性肝疾患患者における血小板減少症の改善>
5.1 血小板数などの臨床検査値や臨床症状、観血的手技の種類から、出血のリスクが高いと考えられる場合に使用すること。
5.2 開腹、開胸、開心、開頭又は臓器切除を伴う観血的手技の場合は、本剤の投与を避けること。有効性及び安全性は確立していない。
<持続性及び慢性免疫性血小板減少症>
5.3 免疫性血小板減少症の発症又は診断後6カ月以上経過した患者に投与すること。
5.4 他の治療にて十分な効果が得られない場合、又は忍容性に問題があると考えられる場合に使用すること。
5.5 血小板数、臨床症状からみて出血リスクが高いと考えられる場合に使用すること。
5.6 免疫性血小板減少症の発症又は診断から6〜12カ月の患者における有効性及び安全性は確立していない。
6. 用法及び用量
<待機的な観血的手技を予定している慢性肝疾患患者における血小板減少症の改善>
通常、成人には、アバトロンボパグとして以下の用量を1日1回、5日間食後に経口投与する。
投与開始前の血小板数が40,000/μL以上50,000/μL未満
40mg
投与開始前の血小板数が40,000/μL未満
60mg
<持続性及び慢性免疫性血小板減少症>
通常、成人には、アバトロンボパグとして初回投与量20mgを1日1回、食後に経口投与する。投与開始後、血小板数、症状に応じて用法・用量を適宜調節する。また、最高投与量は40mgを1日1回とする。
7. 用法及び用量に関連する注意
<待機的な観血的手技を予定している慢性肝疾患患者における血小板減少症の改善>
7.1 本剤の投与は観血的手技の施行予定日の10〜13日前を目安に開始すること。
7.2 本剤を再投与した場合の有効性及び安全性は検討されていない。特に、血小板数が50,000/μL未満に低下していない患者では他の治療法を選択すること。
<持続性及び慢性免疫性血小板減少症>
7.3 本剤は治療上必要最小限の用法・用量で使用すること。
7.4 本剤の用法・用量は下表を参照の上、血小板数に応じて2週間ごとに、血小板数が安定する(少なくとも4週間にわたり用量調節せずに血小板数が50,000/μL以上)まで調節すること。なお、少なくとも2週間は同一用法・用量を維持すること。
ただし、血小板数が50,000/μL未満又は400,000/μL超の場合、1週間に1回、用量調節を行ってもよい。
ただし、血小板数が50,000/μL未満又は400,000/μL超の場合、1週間に1回、用量調節を行ってもよい。
| 用法・用量 | レベル |
| 40mgを1日1回投与 | 6 |
| 40mgを週3回及び20mgを各週の残り4日に投与 | 5 |
| 20mgを1日1回投与 | 4 |
| 20mgを週3回投与 | 3 |
| 20mgを週2回投与又は40mgを週1回投与 | 2 |
| 20mgを週1回投与 | 1 |
| 血小板数 | 調節方法 |
| 50,000/μL未満 | 用量レベルを1段階上げる。 ただし、最高投与量として1日1回40mgを4週間投与しても、臨床上重大な出血リスクを回避できるレベルに血小板数が増加しなかった場合は、本剤の投与を中止するなど、適切な処置を行うこと。 |
| 50,000/μL以上200,000/μL未満 | 現状の用量レベルを維持する。 ただし、出血のリスクを低下できる治療上必要最小限の用法・用量となるよう、適宜減量も考慮すること。 |
| 200,000/μL以上400,000/μL以下 | 用量レベルを1段階下げる。 |
| 400,000/μL超 | 本剤を休薬し、血小板数を週2回測定する。休薬後、血小板数が150,000/μL未満まで減少した場合は、休薬前からの用量レベルを1段階下げて投与を再開する。 ただし、最低投与量として週1回20mgを2週間投与しても血小板数が400,000/μL超の場合は、本剤の投与を中止すること。 |
7.5 本剤投与中は、血小板数が安定するまで(少なくとも4週間にわたり用量調節せずに血小板数が50,000/μL以上)、血小板数を毎週測定すること。血小板数が安定した場合でも4週に1回を目安に血小板数を測定すること。
8. 重要な基本的注意
<効能共通>
<待機的な観血的手技を予定している慢性肝疾患患者における血小板減少症の改善>
8.2 観血的手技の施行前には血小板数が十分に増加していることを確認すること。本剤を投与しても、観血的手技の実施に際し十分な血小板数の増加が得られない場合があるため、必要に応じて血小板輸血の準備をするなど、適切な措置を講じること。
8.3 観血的手技後に血栓症を発現した症例が報告されているため、本剤投与開始後は観察を十分に行うこと。[11.1.1参照]
<持続性及び慢性免疫性血小板減少症>
8.5 本剤は、血液疾患の治療に十分な経験を持つ医師のもとで使用すること。
8.7 本剤の投与中止により血小板減少を認めることがあるため、本剤の中止後4週間程度は血小板数を頻回に測定すること。[11.1.3参照]
8.8 本剤を含むトロンボポエチン受容体作動薬には、骨髄のレチクリン線維の形成及び線維化を進行させる可能性があるので、本剤の投与開始前には、末梢血液像(末梢血塗抹標本)、全血算(赤血球、白血球及び血小板)及び網状赤血球数の検査を行い、全ての血球系の形態異常の有無を十分観察すること。また、本剤投与中は、末梢血液像(末梢血塗抹標本)、全血算(赤血球、白血球及び血小板)及び網状赤血球数の検査を4週に1回を目安に実施し、全ての血球系の形態異常及び血球減少の存否を観察すること。[11.1.2参照]
8.9 トロンボポエチン受容体作動薬には、既存の骨髄異形成症候群等の血液悪性腫瘍を進行させる可能性がある。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 血栓症、血栓塞栓症を有する患者又はそれらの既往歴を有する患者
血栓症又は血栓塞栓症の発現リスクが高くなるおそれがある。臨床試験では除外されている。[8.6参照]
9.1.2 血栓症の発現因子を有する患者
先天性血栓症の発現因子(凝固第V因子ライデン変異・プロトロンビンG20210A変異、抗トロンビン欠損症、プロテインC又はS欠損症など)又は後天性血栓症の発現因子(抗リン脂質抗体症候群など)を有する患者は、血栓症又は血栓塞栓症の発現リスクが高くなるおそれがある。
9.1.3 門脈血流速度が低下している患者
血栓症又は血栓塞栓症の発現リスクが高くなるおそれがある。待機的な観血的手技を予定する血小板減少症を伴う慢性肝疾患患者を対象とした臨床試験では門脈血流速度が10cm/秒未満の患者が除外され、慢性免疫性血小板減少症患者を対象とした臨床試験では門脈圧亢進症患者が除外されている。
9.3 肝機能障害患者
9.3.1 重度の肝機能障害(Child-Pugh分類C)のある患者
投与可否を慎重に判断し、投与する場合は観察を十分に行うこと。
9.5 妊婦
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。ラットで乳汁移行性が認められている2)。
9.7 小児等
小児等を対象とした臨床試験は実施していない。
10. 相互作用
相互作用序文
本剤は主にCYP2C9及びCYP3A4により代謝される。
薬物代謝酵素用語
CYP2C9
薬物代謝酵素用語
CYP3A4
10.2 併用注意
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 血栓症、血栓塞栓症
11.1.2 骨髄線維症(頻度不明)
骨髄線維症があらわれることがある。血球系の形態異常又は血球減少を認めた場合は、本剤の投与を中止すること。また、線維化状態の確認のため骨髄生検・特殊染色等の実施を考慮すること。[8.8参照]
11.1.3 出血(0.3%)
本剤の投与中止後に出血を生じることがある。[8.7参照]
11.2 その他の副作用
| 1-5% | 1%未満 | 頻度不明 | |
| 血液およびリンパ系障害 | 貧血 | ||
| 胃腸障害 | 悪心 | 腹痛 | 歯肉出血 |
| 一般・全身障害および投与部位の状態 | 疲労 | 発熱 | 末梢性浮腫 |
| 免疫系障害 | 過敏症(そう痒、発疹、息詰まり、紅斑、咽頭浮腫、全身性そう痒症、斑状皮疹、顔面腫脹、舌腫脹、蕁麻疹など) | ||
| 感染症および寄生虫症 | 上咽頭炎、上気道感染 | ||
| 傷害、中毒および処置合併症 | 挫傷 | ||
| 代謝および栄養障害 | 低ナトリウム血症 | ||
| 筋骨格系および結合組織障害 | 筋肉痛 | 関節痛 | |
| 神経系障害 | 頭痛 | ||
| 呼吸器、胸郭および縦隔障害 | 鼻出血 | ||
| 皮膚および皮下組織障害 | 点状出血 |
13. 過量投与
13.1 症状
血小板数が過剰に増加し、血栓性又は血栓塞栓性の合併症を起こすおそれがある。
13.2 処置
14. 適用上の注意
14.1 薬剤交付時の注意
14.1.1 ブリスターシートから取り出して服用するよう指導すること。シートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
14.1.2 服用直前にブリスターシートから取り出すよう指導すること。
15. その他の注意
15.1 臨床使用に基づく情報
15.2 非臨床試験に基づく情報
本剤はヒト及びチンパンジー以外のトロンボポエチン受容体に対し親和性を持たず、ヒト及びチンパンジー以外の動物に対して薬理活性を示さない。このため毒性試験において、薬理活性に起因する影響は評価されていない。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
健康成人に本剤20注)、40又は60mgを食後に単回経口投与したときのアバトロンボパグの血漿中濃度推移及び薬物動態パラメータは以下のとおりであった5)。
図1 食後単回経口投与時の血漿中アバトロンボパグ濃度推移(健康成人)(算術平均+標準偏差)
| 投与量(mg) | 例数 | Cmax*1(ng/mL) | Tmax*2(hr) | AUC0-inf*1(ng・hr/mL) | t1/2*1(hr) |
| 20 | 18 | 109(26.9) | 7.00(3.12,12.0) | 3220(831) | 16.4(2.09) |
| 40 | 23 | 208(54.1) | 6.02(3.00,12.0) | 5870(1790) | 16.1(1.96) |
| 60 | 23 | 332(93.3) | 7.00(3.00,24.0) | 9400(2240) | 16.0(1.54) |
16.1.2 反復投与
健康成人に本剤10mg注)を空腹時に1日1回7日間反復経口投与したときのアバトロンボパグの薬物動態パラメータは以下のとおりであった6)。
| 投与量(mg) | 例数 | 投与日 | Cmax*1(ng/mL) | Tmax*2(hr) | AUC0-τ*1(ng・hr/mL) | t1/2*1(hr) |
| 10 | 9 | 1 | 75.2(43.9) | 6.00(4.00,8.00) | 1000(623) | − |
| 7 | 112.8(57.0) | 5.00(3.00,8.00) | 1584(825) | 15.3(2.86) |
<待機的な観血的手技を予定している慢性肝疾患患者における血小板減少症の改善>
血小板減少症を伴う慢性肝疾患患者の成績を用いた母集団薬物動態解析により推定した、本剤40又は60mgを食後に1日1回5日間反復経口投与したときの薬物動態パラメータは以下のとおりであった7)。
| 投与量(mg) | 例数 | Cmax*1(ng/mL) | AUC*1*2(ng・hr/mL) | CL/F*1(L/hr) |
| 40 | 115 | 214.3(42.6) | 3717(62.4) | 7.24(15.6) |
| 60 | 160 | 352.2(47.3) | 4820(85.1) | 7.46(19.6) |
<持続性及び慢性免疫性血小板減少症>
慢性免疫性血小板減少症患者の成績を用いた母集団薬物動態解析により推定した、本剤20mgを食後に1日1回反復経口投与したときの薬物動態パラメータは以下のとおりであった8)。
| 投与量 (mg) | 例数 | Cmax*1 (ng/mL) | AUC0-24*1 (ng・hr/mL) | CL/F*1 (L/hr) |
| 20 | 12 | 263(37.2) | 4991(35) | 5.22(14.6) |
16.2 吸収
16.2.1 食事の影響
健康成人に本剤40又は60mgを単回経口投与したとき、空腹時投与に対する食後投与のCmax及びAUC0-infの幾何平均値の比は、40mgでそれぞれ0.841及び0.922、60mgでそれぞれ0.958及び1.09であった5)。
| 投与量(mg) | 空腹時/食後 | 例数 | Cmax*1(ng/mL) | Tmax*2(hr) | AUC0-inf*1(ng・hr/mL) |
| 40 | 空腹時 | 18 | 239(46.1) | 5.00(3.00,12.0) | 6130(47.9) |
| 食後 | 23 | 201(26.0) | 6.02(3.00,12.0) | 5650(30.5) | |
| 60 | 空腹時 | 18 | 334(53.0) | 5.00(4.00,7.00) | 8420(51.7) |
| 食後 | 23 | 320(28.1) | 7.00(3.00,24.0) | 9160(23.8) |
16.3 分布
アバトロンボパグのヒト血漿蛋白結合率は96%以上であった4)(in vitro)。
16.4 代謝
16.4.1 代謝物
健康成人6例に[14C]-アバトロンボパグ20mg注)を単回経口投与したとき、血漿中に代謝物は検出されなかった。約88%が糞中に排泄され、そのうち33.5%が未変化体、43.8%が4-ヒドロキシ体であった3)(外国人データ)。
16.4.2 代謝酵素
アバトロンボパグは主にCYP2C9及びCYP3A4により代謝された9)(in vitro)。
16.5 排泄
健康成人6例に[14C]-アバトロンボパグ20mg注)を単回経口投与したとき、投与された総放射能の約88%が糞中に、約6%が尿中に排泄された3)(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
血小板減少症を伴う慢性肝疾患患者の成績を用いた母集団薬物動態解析により推定した、腎機能正常患者(CLcr 90mL/min以上)、軽度の腎機能障害患者(CLcr 60mL/min以上90mL/min未満)及び中等度の腎機能障害患者(CLcr 30mL/min以上60mL/min未満)に本剤40又は60mgを食後に1日1回5日間反復経口投与したときの薬物動態パラメータは以下のとおりであった7)。
| 投与量(mg) | CLcr(mL/min) | 例数 | Cmax*1(ng/mL) | AUC*1*2(ng・hr/mL) |
| 40 | 90以上 | 71 | 172(56.8) | 3670(55.4) |
| 60以上90未満 | 26 | 156(69.6) | 3260(76.2) | |
| 30以上60未満 | 14 | 199(64.3) | 4290(62.3) | |
| 60 | 90以上 | 104 | 219(63.8) | 4670(64.8) |
| 60以上90未満 | 37 | 289(77.6) | 6060(83.3) | |
| 30以上60未満 | 15 | 186(112) | 3750(122) |
16.6.2 肝機能障害患者
血小板減少症を伴う慢性肝疾患患者の成績を用いた母集団薬物動態解析により推定した、軽度の肝機能障害患者(Child-Pugh分類A)、中等度の肝機能障害患者(Child-Pugh分類B)及び重度の肝機能障害患者(Child-Pugh分類C)に本剤40又は60mgを食後に1日1回5日間反復経口投与したときの薬物動態パラメータは以下のとおりであった7)。
| 投与量(mg) | Child-Pugh分類 | 例数 | Cmax*1(ng/mL) | AUC*1*2(ng・hr/mL) |
| 40 | A | 63 | 182(61.7) | 3870(59.1) |
| B | 45 | 155(61.0) | 3290(66.5) | |
| C | 7 | 223(30.1) | 5070(29.2) | |
| 60 | A | 92 | 243(79.4) | 5070(81.2) |
| B | 58 | 216(63.2) | 4620(68.2) | |
| C | 7 | 211(49.7) | 4640(60.3) |
16.7 薬物相互作用
16.7.1 In vitro試験
アバトロンボパグはBCRPに対する阻害作用を示し、IC50値は16.4μmol/Lであった10)。
16.7.2 その他の薬剤
(1)イトラコナゾール
健康成人16例を対象にイトラコナゾール(CYP3A4阻害剤)200mgを16日間反復経口投与(投与1日目のみ1日2回投与し、以降は1日1回投与)し、イトラコナゾールの投与7日目に本剤20mg注)を単回経口投与したとき、本剤単独投与時と比較して、本剤のCmaxは7%、AUC0-infは37%増加した11)(外国人データ)。
(2)フルコナゾール
健康成人16例を対象にフルコナゾール(CYP2C9及びCYP3A4阻害剤)400mgを1日1回16日間反復経口投与し、フルコナゾールの投与7日目に本剤20mg注)を単回経口投与したとき、本剤単独投与時と比較して、本剤のCmaxは17%、AUC0-infは116%増加した11)(外国人データ)。
(3)リファンピシン
健康成人16例を対象にリファンピシン(CYP2C9及びCYP3A4誘導剤)600mgを食事1時間前に1日1回16日間反復経口投与し、リファンピシンの投与7日目に本剤20mg注)を単回経口投与したとき、本剤単独投与時と比較して、本剤のCmaxは4%増加、AUC0-infは43%減少した11)(外国人データ)。
(4)シクロスポリン
健康成人36例を対象にシクロスポリン(P-糖蛋白質阻害剤)400mg及び本剤20mg注)を併用して単回経口投与したとき、本剤単独投与時と比較して、本剤のCmaxは34%、AUC0-infは17%減少した12)(外国人データ)。
(5)ベラパミル
健康成人36例を対象にベラパミル(P-糖蛋白質及びCYP3A4阻害剤)240mgを1日1回11日間反復経口投与し、ベラパミルの投与7日目に本剤20mg注)を単回経口投与したとき、本剤単独投与時と比較して、本剤のCmaxは26%、AUC0-infは61%増加した12)(外国人データ)。
注)本剤の承認された用法及び用量は、待機的な観血的手技を予定している慢性肝疾患患者における血小板減少症の改善に対しては40又は60mgの1日1回5日間投与、持続性及び慢性免疫性血小板減少性症に対しては初回投与量として20mgの1日1回投与、最高投与量は40mgの1日1回である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
<待機的な観血的手技を予定している慢性肝疾患患者における血小板減少症の改善>
17.1.1 国際共同第III相試験(E5501-G000-311)
待機的な観血的手技を予定する血小板減少症を伴う慢性肝疾患患者204例(日本人50例を含む)を対象に、多施設共同・ランダム化・二重盲検・プラセボ対照・並行群間比較試験を実施した。本剤[投与前の血小板数が低い(40,000/μL未満)場合は60mg、高い(40,000/μL以上50,000/μL未満)場合は40mg]又はプラセボを1日1回、5日間、食後に経口投与した。血小板数は投与前までの2時点の平均値を用い、いずれの時点も60,000/μL未満であることとされた。また、model for end-stage liver disease(MELD)スコアが24超の患者は除外された。被験者は、治験薬最終投与5〜8日後(治験薬投与開始10〜13日目)に予定する手技を受けることとされた。
投与開始前の血小板数の平均値(最小値−最大値)は、投与前の血小板数が低いコホートでは32,600(12,000-39,700)/μL、血小板数が高いコホートでは44,400(36,000-50,000)/μLであった。実施された観血的手技は、腹水穿刺術、上部消化管内視鏡検査(生検ありの場合を含む)、上部消化管内視鏡下静脈瘤結紮術・硬化療法(生検ありの場合を含む)、大腸内視鏡検査(大腸ポリペクトミー又は生検ありの場合を含む)、肝生検、肝細胞癌に対する化学塞栓療法(TACE)・ラジオ波焼灼術、歯科処置、経頸静脈的肝内門脈大循環短絡術(TIPS)、腹腔鏡下処置及び血管カテーテル手技であった。
有効性の主要評価項目とした血小板輸血及び止血処置を回避した被験者の割合は、投与前の血小板数が低いコホート及び高いコホートのいずれにおいても本剤群でプラセボ群よりも有意に高かった(表8)。以上のように、本剤の有効性のプラセボに対する優越性が検証された。
本剤群及びプラセボ群の血小板数の推移を図2に示す。[8.4参照]
投与開始前の血小板数の平均値(最小値−最大値)は、投与前の血小板数が低いコホートでは32,600(12,000-39,700)/μL、血小板数が高いコホートでは44,400(36,000-50,000)/μLであった。実施された観血的手技は、腹水穿刺術、上部消化管内視鏡検査(生検ありの場合を含む)、上部消化管内視鏡下静脈瘤結紮術・硬化療法(生検ありの場合を含む)、大腸内視鏡検査(大腸ポリペクトミー又は生検ありの場合を含む)、肝生検、肝細胞癌に対する化学塞栓療法(TACE)・ラジオ波焼灼術、歯科処置、経頸静脈的肝内門脈大循環短絡術(TIPS)、腹腔鏡下処置及び血管カテーテル手技であった。
有効性の主要評価項目とした血小板輸血及び止血処置を回避した被験者の割合は、投与前の血小板数が低いコホート及び高いコホートのいずれにおいても本剤群でプラセボ群よりも有意に高かった(表8)。以上のように、本剤の有効性のプラセボに対する優越性が検証された。
本剤群及びプラセボ群の血小板数の推移を図2に示す。[8.4参照]
| 血小板数40,000/μL未満 | 血小板数40,000/μL以上50,000/μL未満 | |||
| プラセボ(N=43) | 本剤60mg(N=70) | プラセボ(N=33) | 本剤40mg(N=58) | |
| 血小板輸血及び止血処置を回避した被験者a),n(%) | 15(34.9) | 48(68.6) | 11(33.3) | 51(87.9) |
| 回避した被験者の割合の95%CIb) | (20.6,49.1) | (55.7,79.4) | (17.2,49.4) | (79.5,96.3) |
| プラセボ群との差(95%CI)c) | − | 33.7(15.8,51.6) | − | 54.6(36.5,72.7) |
| P値(CMH検定)d) | − | 0.0006 | − | <0.0001 |
図2 血小板数の推移(最大の解析対象集団、平均値±標準偏差)[E5501-G000-311試験]
副作用の発現割合は、投与前の血小板数が低いコホート(本剤60mg群)では8.6%(6/70例)、血小板数が高いコホート(本剤40mg群)では7.0%(4/57例)であった。主な副作用(発現割合が1%以上)は、悪心(2.4%)、疲労(1.6%)及び頭痛(1.6%)であった13)。
17.1.2 海外第III相試験(E5501-G000-310)
待機的な観血的手技を予定する血小板減少症を伴う慢性肝疾患患者231例(日本人の登録なし)を対象に、多施設共同・ランダム化・二重盲検・プラセボ対照・並行群間比較試験を実施した。本剤[投与前の血小板数が低い(40,000/μL未満)場合は60mg、高い(40,000/μL以上50,000/μL未満)場合は40mg]又はプラセボを1日1回、5日間、食後に経口投与した。血小板数は投与前までの2時点の平均値を用い、いずれの時点も60,000/μL未満であることとされた。また、model for end-stage liver disease(MELD)スコアが24超の患者は除外された。被験者は、治験薬最終投与5〜8日後(治験薬投与開始10〜13日目)に予定する手技を受けることとされた。
投与開始前の血小板数の平均値(最小値−最大値)は、投与前の血小板数が低いコホートでは31,000(10,000-44,500)/μL、血小板数が高いコホートでは44,500(40,000-50,500)/μLであった。実施された観血的手技は、腹水穿刺術、上部消化管内視鏡検査(生検ありの場合を含む)、上部消化管内視鏡下静脈瘤結紮術・硬化療法(生検ありの場合を含む)、大腸内視鏡検査(大腸ポリペクトミー又は生検ありの場合を含む)、肝生検、肝細胞癌に対するエタノール注入療法・化学塞栓療法(TACE)・ラジオ波焼灼術、歯科処置、経頸静脈的肝内門脈大循環短絡術(TIPS)及び血管カテーテル手技であった。
有効性の主要評価項目とした血小板輸血及び止血処置を回避した被験者の割合は、投与前の血小板数が低いコホート及び高いコホートのいずれにおいても本剤群でプラセボ群よりも有意に高かった(表9)。以上のように、本剤の有効性のプラセボに対する優越性が検証された。
本剤群及びプラセボ群の血小板数の推移を図3に示す。[8.4参照]
投与開始前の血小板数の平均値(最小値−最大値)は、投与前の血小板数が低いコホートでは31,000(10,000-44,500)/μL、血小板数が高いコホートでは44,500(40,000-50,500)/μLであった。実施された観血的手技は、腹水穿刺術、上部消化管内視鏡検査(生検ありの場合を含む)、上部消化管内視鏡下静脈瘤結紮術・硬化療法(生検ありの場合を含む)、大腸内視鏡検査(大腸ポリペクトミー又は生検ありの場合を含む)、肝生検、肝細胞癌に対するエタノール注入療法・化学塞栓療法(TACE)・ラジオ波焼灼術、歯科処置、経頸静脈的肝内門脈大循環短絡術(TIPS)及び血管カテーテル手技であった。
有効性の主要評価項目とした血小板輸血及び止血処置を回避した被験者の割合は、投与前の血小板数が低いコホート及び高いコホートのいずれにおいても本剤群でプラセボ群よりも有意に高かった(表9)。以上のように、本剤の有効性のプラセボに対する優越性が検証された。
本剤群及びプラセボ群の血小板数の推移を図3に示す。[8.4参照]
| 血小板数40,000/μL未満 | 血小板数40,000/μL以上50,000/μL未満 | |||
| プラセボ(N=48) | 本剤60mg(N=90) | プラセボ(N=34) | 本剤40mg(N=59) | |
| 血小板輸血及び止血処置を回避した被験者a),n(%) | 11(22.9) | 59(65.6) | 13(38.2) | 52(88.1) |
| 回避した被験者の割合の95%CIb) | (11.0,34.8) | (55.7,75.4) | (21.9,54.6) | (79.9,96.4) |
| プラセボ群との差(95%CI)c) | − | 42.6(27.2,58.1) | − | 49.9(31.6,68.2) |
| P値(CMH検定)d) | − | <0.0001 | − | <0.0001 |
図3 血小板数の推移(最大の解析対象集団、平均値±標準偏差)[E5501-G000-310試験]
副作用の発現割合は、投与前の血小板数が低いコホート(本剤60mg群)では13.5%(12/89例)、血小板数が高いコホート(本剤40mg群)では6.9%(4/58例)であった。主な副作用(発現割合が1%以上)は、頭痛(2.0%)、悪心(1.4%)、疲労(1.4%)、骨痛(1.4%)及び浮動性めまい(1.4%)であった14)。
<持続性及び慢性免疫性血小板減少症>
17.1.3 国内第III相試験(AVA-ITP-307)
過去の治療で十分な効果が得られなかった成人免疫性血小板減少症患者19例を対象に、多施設共同・非盲検・非対照試験を実施した。本試験は、診断後12カ月以上で、投与前までの2時点の平均血小板数が30,000/μL未満の免疫性血小板減少症患者を対象とした。免疫性血小板減少症に対する前治療は、副腎皮質ステロイド、トロンボポエチン受容体作動薬、リツキシマブ、静注用人免疫グロブリン製剤及び免疫抑制剤であった。初回投与量として本剤20mgを1日1回食後に投与し、2週間ごとに血小板数に応じて用量調節を行い、20mgを週1回、20mgを週2回又は40mgを週1回、20mgを週3回、20mgを1日1回、40mgを週3回及び20mgを週4回、40mgを1日1回のいずれかで投与した。
有効性の主要評価項目である血小板反応の累積週数(コア期26週間のうち、救済療法を実施せずに血小板数が50,000/μL以上となった週数)の95%信頼区間の下限値は、事前に規定した閾値である8.02週を上回った(表10)。26週の投与期間における血小板数の推移を図4に示す。
有効性の主要評価項目である血小板反応の累積週数(コア期26週間のうち、救済療法を実施せずに血小板数が50,000/μL以上となった週数)の95%信頼区間の下限値は、事前に規定した閾値である8.02週を上回った(表10)。26週の投与期間における血小板数の推移を図4に示す。
| 主要評価項目 | アバトロンボパグ (N=19) |
| 平均値(標準偏差) | 13.47(9.002) |
| 95%信頼区間 | 9.13,17.80 |
| 中央値 | 16.57 |
| 最小値,最大値 | 0.0,25.1 |
図4 血小板数の推移(最大の解析対象集団、中央値(第1四分位数、第3四分位数))[コア期(AVA-ITP-307試験)]
図中に記載された数値は測定時点の解析対象例数であり、投与期間中に試験を中止した患者は、中止した時点以降の血小板数の評価から除外された。
副作用の発現割合は、15.8%(3/19例)であった。主な副作用(発現割合が5%以上)は、白血球増加症(5.3%)、動悸(5.3%)、血圧上昇(5.3%)、頭痛(5.3%)及び蕁麻疹(5.3%)であった15)。
18. 薬効薬理
18.1 作用機序
アバトロンボパグは、経口投与可能な低分子のトロンボポエチン受容体作動薬であり、造血前駆細胞から巨核球の増殖及び分化を促進し、血小板数を増加させる。アバトロンボパグは、トロンボポエチンと競合することなくトロンボポエチン受容体に結合し、血小板産生を促進する。
18.2 血小板造血作用
18.2.1 ヒト臍帯血CD34陽性細胞に対してアバトロンボパグは遺伝子組換えヒトトロンボポエチンと同程度の巨核球コロニー形成能を示した16)。
18.2.2 ヒトの造血幹細胞(胎児肝臓由来CD34陽性細胞)を移植したNOD/SCIDマウスにアバトロンボパグを反復経口投与することにより、用量依存的にヒト血小板数が増加した17)。
18.3 血小板機能
本剤40又は60mgを食後に1日1回5日間反復経口投与した慢性肝疾患患者20例から採取した血小板では、アデノシン二リン酸又はトロンビン受容体アゴニストペプチドの添加の有無によらず、血小板活性化に明らかな影響は認められなかった18)。
19. 有効成分に関する理化学的知見
19.1. アバトロンボパグマレイン酸塩
| 一般的名称 | アバトロンボパグマレイン酸塩 |
|---|---|
| 一般的名称(欧名) | Avatrombopag Maleate |
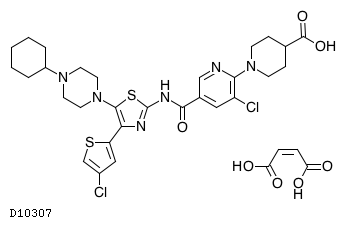
| 化学名 | 1-(3-Chloro-5-{[4-(4-chlorothiophen-2-yl)-5-(4-cyclohexylpiperazin-1-yl)-1,3-thiazol-2-yl]carbamoyl}pyridin-2-yl)piperidine-4-carboxylic acid monomaleate |
| 分子式 | C29H34Cl2N6O3S2・C4H4O4 |
| 分子量 | 765.73 |
| 融点 | 218℃(マレイン酸の離脱) |
| 物理化学的性状 | 白色の粉末 1,3-ジメチル-2-イミダゾリジノン、ジメチルスルホキシド、N-メチルピロリドンに溶けやすく、メタノール、エタノール(99.5)に溶けにくく、水、アセトニトリル、アセトン、酢酸エチル、ヘキサン、tert-ブチルメチルエーテルにほとんど溶けない。 |
| 分配係数 | (logD):>4.0(pH3〜9、1-オクタノール/水) |
| KEGG DRUG |
|
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
10錠[10錠(ブリスターシート)×1]/箱
15錠[15錠(ブリスターシート)×1]/箱
30錠[15錠(ブリスターシート)×2]/箱
23. 主要文献
- 社内資料:胚・胎児発生に関する試験(2023年3月27日承認、申請資料概要2.6.6.6)
- 社内資料:乳汁中排泄(2023年3月27日承認、申請資料概要2.6.4.6.3)
- 社内資料:ヒトマスバランス試験(501-PK-901試験)(2023年3月27日承認、申請資料概要2.7.2.2.1.3)
- 社内資料:in vitro血漿蛋白結合(2023年3月27日承認、申請資料概要2.6.4.4.2)
- 社内資料:国際共同第1相試験(E5501-A001-018試験)(2023年3月27日承認、申請資料概要2.7.2.2.2.2)
- 社内資料:日本人、中国人及び白人での薬物動態及び薬力学試験(E5501-A001-006)(2023年3月27日承認、申請資料概要2.7.2.2.2.1)
- 社内資料:母集団薬物動態/薬力学解析報告書(CPMS-E5501-005R-v1.0)(2023年3月27日承認、申請資料概要2.7.2.2.6.2)
- 社内資料:免疫性血小板減少症を有する日本人被験者の母集団PK及びPK/PD解析(2025年8月25日承認、申請資料概要2.7.2.2.6.3)
- 社内資料:in vitro薬物代謝(2023年3月27日承認、申請資料概要2.6.4.5.1)
- 社内資料:排出トランスポーターとの相互作用(2023年3月27日承認、申請資料概要2.6.4.8.1)
- 社内資料:薬物相互作用試験(E5501-G000-019試験)(2023年3月27日承認、申請資料概要2.7.2.2.4.2)
- 社内資料:薬物相互作用試験(E5501-G000-008試験)(2023年3月27日承認、申請資料概要2.7.2.2.4.1)
- 社内資料:国際共同第3相試験(E5501-G000-311試験)(2023年3月27日承認、申請資料概要2.7.6.19)
- 社内資料:海外共同第3相試験(E5501-G000-310試験)(2023年3月27日承認、申請資料概要2.7.6.18)
- 社内資料:国内第3相試験(AVA-ITP-307試験)(2025年8月25日承認、申請資料概要2.7.6.2)
- 社内資料:in vitro薬理試験(2023年3月27日承認、申請資料概要2.6.2.2.1)
- 社内資料:in vivo薬理試験(2023年3月27日承認、申請資料概要2.6.2.2.2)
- 社内資料:血小板機能評価報告書(2023年3月27日承認、申請資料概要2.7.3.3.2.7)
24. 文献請求先及び問い合わせ先
文献請求先
旭化成セラピューティクス株式会社
くすり相談窓口
〒100-0006
東京都千代田区有楽町一丁目1番2号
電話:フリーダイヤル 0120-114-936 (9:00〜17:45/土日祝、休業日を除く)
製品情報問い合わせ先
旭化成セラピューティクス株式会社
くすり相談窓口
〒100-0006
東京都千代田区有楽町一丁目1番2号
電話:フリーダイヤル 0120-114-936 (9:00〜17:45/土日祝、休業日を除く)
25. 保険給付上の注意
25.1 本製剤の効能又は効果に関連する注意において、「開腹、開胸、開心、開頭又は臓器切除を伴う観血的手技の場合は、本剤の投与を避けること。」とされていることから、このような症例には使用しないこと。また、観血的手技の名称及び実施予定年月日を診療報酬明細書の摘要欄に記入すること。
26. 製造販売業者等
26.1 製造販売元
Swedish Orphan Biovitrum Japan株式会社
東京都港区虎ノ門二丁目6番1号
26.2 発売元
旭化成セラピューティクス株式会社
東京都千代田区有楽町一丁目1番2号