医薬品情報
| 総称名 | パルモディア |
|---|---|
| 一般名 | ペマフィブラート |
| 欧文一般名 | Pemafibrate |
| 製剤名 | ペマフィブラート徐放錠 |
| 薬効分類名 | 高脂血症治療剤 |
| 薬効分類番号 | 2183 |
| ATCコード | C10AB12 |
| KEGG DRUG |
D10711
ペマフィブラート
|
| KEGG DGROUP |
DG01946
脂質低下薬
|
| JAPIC | 添付文書(PDF) |
添付文書情報2026年2月 改訂(第6版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| パルモディアXR錠0.2mg | PARMODIA XR TABLETS 0.2mg | 興和 | 2183007G1020 | 51円/錠 | 処方箋医薬品注) |
| パルモディアXR錠0.4mg | PARMODIA XR TABLETS 0.4mg | 興和 | 2183007G2027 | 94.4円/錠 | 処方箋医薬品注) |
2. 禁忌
次の患者には投与しないこと
4. 効能または効果
高脂血症(家族性を含む)
5. 効能または効果に関連する注意
5.1 LDL-コレステロールのみが高い高脂血症に対し、第一選択薬とはしないこと。(HMG-CoA還元酵素阻害薬の使用が適さない患者を除く)。
5.2 LDL-コレステロールの低下を目的として投与する場合には、以下に注意すること。
5.2.1 LDL-コレステロールが高くトリグリセライドが正常値であり、以下に示すHMG-CoA還元酵素阻害薬による治療が適さない高脂血症に使用すること。
・副作用の既往等によりHMG-CoA還元酵素阻害薬の使用が困難な患者
・HMG-CoA還元酵素阻害薬の使用が禁忌とされる患者
5.2.2 臨床試験に組み入れられた患者のLDL-コレステロール値及びトリグリセライド値について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、適応患者を選択すること。[17.1.3参照]
5.2.3 家族性高コレステロール血症のうちホモ接合体については使用経験がないことから、治療上やむを得ないと判断される場合のみ、LDLアフェレーシス等の非薬物療法の補助として本剤の適用を考慮すること。
5.3 適用の前に十分な検査を実施し、高脂血症の診断が確立した患者に対してのみ本剤の適用を考慮すること。
6. 用法及び用量
通常、成人にはペマフィブラートとして1回0.2mgを1日1回経口投与する。ただし、トリグリセライド又はLDL-コレステロール高値の程度により、1回0.4mgを1日1回まで増量できる。
7. 用法及び用量に関連する注意
8. 重要な基本的注意
8.1 あらかじめ高脂血症治療の基本である食事療法を行い、更に運動療法や、高血圧・喫煙等の虚血性心疾患のリスクファクターの軽減も十分考慮すること。
8.2 投与中は血清脂質値を定期的に検査し、本剤の効果が認められない場合には漫然と投与せず、中止すること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 胆石の既往歴のある患者
胆石形成が報告されている。
9.2 腎機能障害患者
9.2.1 eGFRが30mL/min/1.73m2未満の腎機能障害のある患者
9.2.2 腎機能に関する臨床検査値に異常が認められる患者
9.3 肝機能障害患者
9.3.1 重篤な肝障害、Child-Pugh分類B又はCの肝硬変のある患者あるいは胆道閉塞のある患者
9.3.2 肝障害のある患者(Child-Pugh分類Aの肝硬変のある患者等)又は肝障害の既往歴のある患者(9.3.1に該当する患者を除く)
9.5 妊婦
妊婦又は妊娠している可能性のある女性には投与しないこと。[2.4参照]
9.6 授乳婦
授乳しないことが望ましい。動物実験(ラット)で乳汁中への移行が報告されている。
9.7 小児等
小児等を対象とした有効性及び安全性を指標とした臨床試験は実施していない。
9.8 高齢者
副作用発現に留意し、経過を十分に観察しながら慎重に投与すること。一般に生理機能が低下している。
10. 相互作用
相互作用序文
本剤は、主としてCYP2C8、CYP2C9、CYP3Aにより代謝される。また、本剤は、OATP1B1、OATP1B3の基質となる。
薬物代謝酵素用語
CYP2C8
薬物代謝酵素用語
CYP2C9
薬物代謝酵素用語
CYP3A
薬物代謝酵素用語
OATP1B1
薬物代謝酵素用語
OATP1B3
10.1 併用禁忌
10.2 併用注意
| HMG-CoA還元酵素阻害薬 プラバスタチンナトリウム シンバスタチン フルバスタチンナトリウム等 [9.2.2、11.1.1参照] | 急激な腎機能悪化を伴う横紋筋融解症があらわれやすい。自覚症状(筋肉痛、脱力感)の発現、CK上昇、血中及び尿中ミオグロビン上昇並びに血清クレアチニン上昇等の腎機能の悪化を認めた場合は直ちに投与を中止すること。 | 危険因子:腎機能に関する臨床検査値に異常が認められる患者 |
| クロピドグレル硫酸塩 [16.7.1参照] | 併用する場合には本剤投与の適否及び本剤の増量の必要性を慎重に判断すること。併用により本剤の血漿中濃度が上昇するおそれがある。 | 左記薬剤のCYP2C8及びOATP1B1の阻害作用によると考えられる。 |
| クラリスロマイシン HIVプロテアーゼ阻害剤 リトナビル等 [16.7.1参照] | 併用する場合には本剤投与の適否及び本剤の増量の必要性を慎重に判断すること。併用により本剤の血漿中濃度が上昇するおそれがある。 | 左記薬剤のCYP3A、OATP1B1及びOATP1B3の阻害作用によると考えられる。 |
| フルコナゾール [16.7.1参照] | 併用により本剤の血漿中濃度が上昇するおそれがある。 | 左記薬剤のCYP2C9及びCYP3Aの阻害によると考えられる。 |
| 陰イオン交換樹脂 コレスチラミン コレスチミド | 本剤の血漿中濃度が低下する可能性があるので、併用する場合には、可能な限り間隔をあけて投与することが望ましい。 | 同時投与により本剤が左記薬剤に吸着され吸収が低下する可能性がある。 |
| 強いCYP3A誘導剤 カルバマゼピン フェノバルビタール フェニトイン セイヨウオトギリソウ(セント・ジョーンズ・ワート)含有食品等 | 本剤の血漿中濃度が低下し、本剤の効果が減弱するおそれがある。 | 左記薬剤の強いCYP3Aの誘導作用により、本剤の代謝が促進されると考えられる。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 横紋筋融解症(頻度不明)
11.1.2 肝機能障害、黄疸(いずれも頻度不明)[8.3参照]
11.2 その他の副作用
| 0.5%以上 | 0.1〜0.5%未満 | 頻度不明 | |
| 肝臓 | ALT上昇 | 胆石症、肝機能異常、AST上昇 | |
| 筋肉 | CK上昇、筋肉痛 | 血中ミオグロビン増加 | |
| 皮膚 | 発疹 | そう痒 | |
| その他 | 糖尿病(悪化を含む) | グリコヘモグロビン増加、低比重リポ蛋白増加、血中尿酸増加 |
14. 適用上の注意
14.1 薬剤交付時の注意
14.1.1 PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
14.1.2 本剤は徐放性製剤であるため、砕いたり、すりつぶしたりしないで、そのままかまずに服用するよう指導すること。砕いたり、すりつぶしたりして服用すると、本剤の徐放性が失われ、薬物動態が変わるおそれがある。
15. その他の注意
15.2 非臨床試験に基づく情報
マウスのがん原性試験(0.075mg/kg/日以上)で肝細胞癌及び肝細胞腺腫の発現頻度の増加が認められた。ラットのがん原性試験(雄0.3mg/kg/日以上、雌1mg/kg/日以上)で肝細胞癌及び肝細胞腺腫、膵臓腺房細胞癌、膵臓腺房細胞腺腫、精巣ライディッヒ細胞腺腫並びに甲状腺濾胞上皮細胞腺腫の発現頻度の増加が認められた。
16. 薬物動態
16.1 血中濃度
16.1.1 反復投与
トリグリセライド(TG)高値の脂質異常症患者に本剤0.4mg/日を1日1回又はペマフィブラート即放性製剤(IR錠)0.2mg/日を1日2回に分けて食前又は食後に4週間反復経口投与した(2期クロスオーバー)。本剤投与4週時の血漿中ペマフィブラート濃度推移は次図のとおりであり、薬物動態パラメータは次表のとおりであった。投与4週時の食前投与に対する食後投与のCmax及びAUC0-τの幾何平均値の比[90%信頼区間]は、1.124[0.840,1.503]及び1.097[0.879,1.370]であった。
なお、ペマフィブラートの1日用量を同等に補正したIR錠0.2mg/日投与4週時に対する本剤0.4mg/日投与4週時のAUC0-τの幾何平均値の比[90%信頼区間]は、食前投与時と食後投与時でそれぞれ0.863[0.797,0.934]、0.870[0.788,0.960]であった1)。
なお、ペマフィブラートの1日用量を同等に補正したIR錠0.2mg/日投与4週時に対する本剤0.4mg/日投与4週時のAUC0-τの幾何平均値の比[90%信頼区間]は、食前投与時と食後投与時でそれぞれ0.863[0.797,0.934]、0.870[0.788,0.960]であった1)。
図 TG高値の脂質異常症患者における本剤0.4mg/日食前又は食後反復経口投与4週時の血漿中ペマフィブラート濃度推移
| \ | Cmax(ng/mL) | AUC0-τ(ng・h/mL) | tmax(h) | t1/2(h) |
| 食前投与 | 3.1283[70.6] n=20 | 22.7233[61.8] n=20 | 3.00[1.5,14.0] n=20 | 5.549[43.6] n=17 |
| 食後投与 | 3.5149[59.3] n=19 | 24.9334[37.7] n=19 | 8.00[3.0,11.9] n=19 | 4.185[23.6] n=17 |
16.2 吸収
16.2.1 バイオアベイラビリティ
健康成人男性8例にペマフィブラート即放性製剤(IR錠)0.2mgを単回経口投与したとき、IR錠の絶対バイオアベイラビリティは61.5%であった2)(外国人データ)。
16.3 分布
16.3.1 蛋白結合率
ペマフィブラートのヒト血漿蛋白結合率は99%以上であった3)(in vitro)。
16.4 代謝
16.4.1 血漿中代謝物
健康成人男性8例に14C-ペマフィブラート0.8mg注1)を単回経口投与したとき、主な血漿中代謝物はベンジル位酸化体及びジカルボン酸体のグルクロン酸抱合体とN-脱アルキル体の混合物であった2)(外国人データ)。
16.4.2 代謝酵素
ペマフィブラートは、CYP2C8、CYP2C9、CYP3A4、CYP3A7、UGT1A1、UGT1A3及びUGT1A8の基質である3)(in vitro)。
16.5 排泄
16.5.1 尿中及び糞中排泄率
健康成人男性7例に14C-ペマフィブラート0.8mg注1)を単回経口投与したとき、投与216時間後までの尿及び糞中へ投与放射能の14.53%及び73.29%が排泄された2)(外国人データ)。
16.5.2 トランスポーター
ペマフィブラートは、P-gp、BCRP、OATP1A2、OATP1B1、OATP1B3、OCT2及びNTCPの基質である3)(in vitro)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
(1)腎機能障害患者(軽度、中等度、高度又は末期腎不全)30例に、ペマフィブラート即放性製剤(IR錠)0.2mgを単回経口投与したときの腎機能正常者に対する薬物動態パラメータの比は次表のとおりであり、腎機能正常者群と比較して、腎機能障害患者群では曝露の増加が認められたが、腎機能障害の程度に依存した曝露の増加は認められなかった4)。[7.、9.2.1参照]
| \ | Cmax | AUC0-t |
| 軽度腎機能障害患者群 [50≦Ccr<80mL/min](n=8) | 1.644[1.155,2.342] | 1.629[1.161,2.287] |
| 中等度腎機能障害患者群 [30≦Ccr<50mL/min](n=8) | 1.093[0.767,1.556] | 1.154[0.822,1.620] |
| 高度腎機能障害患者群 [Ccr<30mL/min](n=7) | 1.545[1.072,2.228] | 1.296[0.913,1.841] |
| 末期腎不全患者群 [血液透析で治療中](n=7) | 1.258[0.872,1.813] | 1.607[1.131,2.282] |
(2)腎機能障害(高度腎機能障害(eGFR<30mL/min/1.73m2又は透析)及び軽度〜中等度腎機能障害(30≦eGFR<60mL/min/1.73m2))を有するTG高値の脂質異常症患者に、ペマフィブラート即放性製剤(IR錠)0.2mg/日を1日2回に分けて朝夕12週間反復経口投与した。12週時におけるAUC0-τについて、軽度〜中等度腎機能障害群(対照群)に対する高度腎機能障害群の幾何平均値の比及びその90%信頼区間は次表のとおりであり、高度腎機能障害患者においても曝露の増加は認められなかった。
| \ | AUC0-τの幾何平均値の比[90%信頼区間] |
| 高度腎機能障害群 [eGFR<30mL/min/1.73m2又は透析] | 0.9177[0.6198,1.3587] |
なお、血漿中薬物動態パラメータは次表のとおりであった5)。[7.、9.2.1参照]
| \ | Cmax(ng/mL) | AUC0-τ(ng・h/mL) |
| 軽度〜中等度腎機能障害患者 [30≦eGFR<60mL/min/1.73m2](n=7) | 2.4483±0.9535 | 8.6994±4.0397 |
| 高度腎機能障害患者 [eGFR<30mL/min/1.73m2](n=4) | 2.0508±0.6588 | 7.4130±3.9548 |
| 高度腎機能障害患者 [透析](n=4) | 1.8798±0.5728 | 8.4470±3.3054 |
16.6.2 脂肪肝患者及び肝硬変患者
脂肪肝患者及び肝硬変患者24例に、ペマフィブラート即放性製剤(IR錠)0.2mgを単回経口投与したときの肝機能正常者に対する薬物動態パラメータの比は次表のとおりであり、肝機能正常者群と比較して、脂肪肝患者群及び肝硬変患者群では曝露の増加が認められた6)。[2.2、9.3.1、9.3.2参照]
| \ | Cmax | AUC0-t |
| 脂肪肝患者群 (n=10) | 1.198[0.819,1.750] | 1.194[0.836,1.707] |
| 軽度の肝硬変患者群 Child-Pugh分類A(n=8) | 2.329[1.561,3.475] | 2.076[1.425,3.026] |
| 中等度の肝硬変患者群 Child-Pugh分類B(n=6) | 3.882[2.520,5.980] | 4.191[2.790,6.294] |
16.7 薬物相互作用
16.7.1 シクロスポリン、リファンピシン、クロピドグレル、クラリスロマイシン、フルコナゾールとの併用
健康成人にペマフィブラート即放性製剤(IR錠)と各種薬剤を併用投与したとき、薬物動態パラメータ等への影響は次表のとおりであった7)(外国人データ)。[2.5、10.1、10.2参照]
| 併用薬 | 併用薬投与量 | IR錠投与量注2) | 測定対象 | 幾何平均値の比[90%信頼区間] (併用投与時/単独投与時) | |
| Cmax | AUC0-inf | ||||
| シクロスポリン | 600mg 単回 | 0.4mg 単回 | 本薬 | 8.9644[7.5151,10.6931] n=14 | 13.9947[12.6175,15.5223] n=12 |
| リファンピシン | 600mg 単回 | 0.4mg 単回 | 本薬 | 9.4336[8.3626,10.6419] n=20 | 10.9009[9.9154,11.9844] n=17 |
| 600mg/日 1日1回 10日間 単独投与 | 0.4mg 単回 単独投与 | 本薬 | 0.3792a)[0.3378,0.4257] n=20 | 0.2221a)[0.2065,0.2389] n=16 | |
| クロピドグレル | 300mg 単回 4日目 | 0.4mg 単回 4日目 | 本薬 | 1.4855[1.3915,1.5858] n=20 | 2.3728[2.2473,2.5052] n=20 |
| 75mg/日 1日1回 5日間 5〜9日目 | 0.4mg 単回 7日目 | 本薬 | 1.3415[1.2583,1.4302] n=20 | 2.0876[1.9811,2.1998] n=20 | |
| クラリスロマイシン | 1,000mg/日 1日2回 8日間 | 0.4mg 単回 | 本薬 | 2.4246[2.1632,2.7174] n=18 | 2.0975[1.9158,2.2964] n=17 |
| フルコナゾール | 400mg/日 1日1回 11日間 | 0.4mg 単回 | 本薬 | 1.4409[1.2899,1.6096] n=19 | 1.7891[1.6638,1.9239] n=17 |
16.7.2 HMG-CoA還元酵素阻害薬との併用
健康成人男性にペマフィブラート即放性製剤(IR錠)とHMG-CoA還元酵素阻害薬を併用投与したとき、薬物動態パラメータへの影響は次表のとおりであった8)(外国人データを含む)。
| 併用薬 | 併用薬投与量 | IR錠投与量 | 測定対象 | 幾何平均値の比[90%信頼区間] (併用投与時/単独投与時) | |
| Cmax | AUC0-τ | ||||
| アトルバスタチン | 20mg/日 1日1回 7日間 | 0.4mg/日 1日2回 7日間 | 本薬 (n=18) | 1.166[1.069,1.272] | 1.098[1.016,1.187] |
| アトルバスタチン (n=18) | 1.032[0.960,1.109] | 0.934[0.851,1.024] | |||
| o-ヒドロキシアトルバスタチン (n=18) | 0.875[0.826,0.927] | 0.784[0.736,0.836] | |||
| シンバスタチン | 20mg/日 1日1回 7日間 | 0.4mg/日 1日2回 7日間 | 本薬 (n=18) | 1.230[1.090,1.388] | 1.125[0.997,1.270] |
| シンバスタチン (n=19) | 0.858[0.660,1.114] | 0.846[0.722,0.992] | |||
| シンバスタチンオープンアシド体 (n=19) | 0.626[0.541,0.725] | 0.405[0.345,0.475] | |||
| ピタバスタチン | 4mg/日 1日1回 7日間 | 0.4mg/日 1日2回 7日間 | 本薬 (n=18) | 1.061[0.970,1.160] | 1.122[1.041,1.209] |
| ピタバスタチン (n=18) | 1.011[0.973,1.050] | 1.036[1.007,1.066] | |||
| プラバスタチン | 20mg/日 1日1回 7日間 | 0.4mg/日 1日2回 7日間 | 本薬 (n=18) | 1.058[0.964,1.162] | 1.057[1.013,1.102] |
| プラバスタチン (n=18) | 1.107[0.908,1.351] | 1.065[0.922,1.231] | |||
| フルバスタチン | 60mg/日 1日1回 7日間 | 0.4mg/日 1日2回 7日間 | 本薬 (n=18) | 1.181[1.080,1.290] | 1.207[1.144,1.274] |
| フルバスタチン (n=18) | 0.989[0.790,1.239] | 1.151[1.057,1.253] | |||
| ロスバスタチン | 20mg/日 1日1回 7日間 | 0.4mg/日 1日2回 7日間 | 本薬 (外国人,n=24) | 1.106[1.048,1.167] | 1.110[1.046,1.177] |
| ロスバスタチン (外国人,n=24) | 1.092[1.016,1.174] | 1.025[0.964,1.091] | |||
16.7.3 その他の薬剤
ペマフィブラート即放性製剤(IR錠)とジゴキシン、ワルファリンをそれぞれ併用投与したとき、ペマフィブラートはこれらの薬剤の薬物動態に影響を与えなかった7)(外国人データ)。
注1)本剤の承認された用法及び用量は、「通常、成人にはペマフィブラートとして1回0.2mgを1日1回経口投与する。ただし、トリグリセライド又はLDL-コレステロール高値の程度により、1回0.4mgを1日1回まで増量できる。」である。
注2)ペマフィブラート即放性製剤(IR錠)の承認された用法及び用量は、「通常、成人にはペマフィブラートとして1回0.1mgを1日2回朝夕に経口投与する。なお、年齢、症状に応じて適宜増減するが、最大用量は1回0.2mgを1日2回までとする。」である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国内第III相検証試験
TG高値の脂質異常症患者に本剤0.2mg/日又は0.4mg/日を1日1回、ペマフィブラート即放性製剤(IR錠)0.2mg/日を1日2回に分けて12週間投与したとき、空腹時血清TGのベースラインからの変化率は次表のとおりであり、本剤0.2mg/日群及び0.4mg/日群のIR錠0.2mg/日群に対する非劣性が認められた。
| \ | 本剤0.2mg/日(1日1回) | 本剤0.4mg/日(1日1回) | IR錠0.2mg/日(1日2回) |
| ベースラインa) | 338.6±117.0(117) | 355.0±157.5(119) | 354.2±142.3(117) |
| 4週時a) | 186.6±97.4(116) | 177.1±103.4(119) | 168.3±70.4(116) |
| 8週時a) | 180.2±76.0(116) | 170.6±75.9(119) | 179.2±102.5(117) |
| 12週時a) | 192.1±90.6(115) | 166.5±81.6(118) | 173.5±80.7(116) |
| 変化率(%)b) | −43.80[−47.20,−40.39] | −48.00[−51.37,−44.63] | −48.00[−51.40,−44.60] |
| IR錠0.2mg/日群との差(%)b) | 4.20[−0.62,9.02] | 0.00[−4.79,4.79] | − |
また、各評価時点におけるLDL-コレステロールの推移は次表のとおりであった。
| \ | 本剤0.2mg/日 | 本剤0.4mg/日 | IR錠0.2mg/日 |
| ベースライン | 139.0±43.6(117) | 132.3±35.5(119) | 137.3±42.2(117) |
| 4週時 | 139.4±34.9(116) | 140.0±36.2(119) | 141.1±39.3(116) |
| 8週時 | 139.4±39.1(116) | 140.7±37.8(119) | 140.8±40.7(117) |
| 12週時 | 142.3±33.4(115) | 142.3±34.4(118) | 140.9±38.5(116) |
17.1.2 国内第III相長期投与試験
TG高値の脂質異常症患者に本剤0.2mg/日(効果不十分の場合、12週以降に0.4mg/日に増量)を1日1回朝又は夕に52週間投与したとき、最終評価時(52週時又は中止時)及びその直前の時点における空腹時血清TGのベースライン(264.0±109.2mg/dL(平均値±標準偏差)、n=121)からの変化率の平均値[95%信頼区間]は、朝投与群、夕投与群でそれぞれ−44.82%[−49.70,−39.94](n=61)、−46.61%[−51.34,−41.88](n=60)であった。朝投与群における夕投与群との最小二乗平均値の差[95%信頼区間]は3.03%[−3.55,9.62]であった。
また、各評価時点におけるLDL-コレステロールの推移は次表のとおりであった。
また、各評価時点におけるLDL-コレステロールの推移は次表のとおりであった。
| \ | 本剤 | |
| 朝投与群 | 夕投与群 | |
| ベースライン | 120.4±30.7(61) | 130.1±40.9(60) |
| 4週時 | 122.2±28.2(61) | 123.7±41.0(59) |
| 16週時 | 125.5±24.3(61) | 121.4±31.3(56) |
| 28週時 | 123.4±24.4(61) | 119.4±31.9(56) |
| 40週時 | 127.6±30.6(61) | 122.0±34.0(56) |
| 52週時 | 121.2±26.4(59) | 120.1±35.9(55) |
17.1.3 国内第III相検証試験
LDL-コレステロールが高く注1)TGが正常値注2)でスタチン不耐注3)の脂質異常症患者にプラセボ、本剤0.2mg/日又は0.4mg/日を1日1回12週間投与したとき、LDL-コレステロールのベースラインからの変化率は次表のとおりであり、本剤0.2mg/日群及び0.4mg/日群のプラセボ群に対する優越性が認められた。
| \ | プラセボ | 本剤0.2mg/日 | 本剤0.4mg/日 |
| ベースラインa) | 184.82±36.51(24) | 174.27±35.78(21) | 171.10±27.55(23) |
| 4週時a) | 182.57±34.34(24) | 138.51±39.43(21) | 124.07±17.98(23) |
| 8週時a) | 181.33±31.27(24) | 141.49±47.15(20) | 126.82±23.14(21) |
| 12週時a) | 181.70±30.72(24) | 142.60±47.93(18) | 134.18±25.96(20) |
| 変化率(%)b) | −0.35[−4.17,3.46] | −20.00[−24.09,−15.92] | −24.82[−28.76,−20.88] |
| プラセボ群との差(%)b) | − | −19.65[−25.26,−14.04]p<0.001c) | −24.47[−30.00,−18.94]p<0.001c) |
本剤投与による副作用発現割合は、0.2mg/日群、0.4mg/日群でそれぞれ21.7%(5/23例)、16.7%(4/24例)であった。副作用は0.2mg/日群で血中クレアチンホスホキナーゼ増加、頭痛、末梢性ニューロパチー/色素沈着障害、下痢、腱痛が各1例(4.3%)、0.4mg/日群でリポ蛋白(a)増加、尿潜血陽性/胃腸炎、血小板数増加、筋肉痛が各1例(4.2%)ずつ認められた11)。[5.2.2参照]
注1)LDL-Cが、動脈硬化性疾患予防ガイドライン2022年版に基づく各リスク区分において、以下の基準を満たす。
・一次予防の低リスクに該当する場合、160mg/dL以上
・一次予防の中リスクに該当する場合、140mg/dL以上
・一次予防の高リスクに該当する場合、120mg/dL以上
・二次予防に該当する場合、120mg/dL以上
注2)空腹時TGが150mg/dL未満
注3)スタチン(HMG-CoA還元酵素阻害薬)服用に伴って見られる有害事象により、服用者の日常生活に許容困難な障害が生じ、その結果スタチンの服薬中断や減量に至るもの
17.2 製造販売後調査等
17.2.1 2型糖尿病を合併した脂質異常症患者を対象とした国際共同臨床試験
軽度〜中等度のTG高値かつHDL-コレステロール低値を示す2型糖尿病を合併した脂質異常症患者10,497例(日本人305例を含む)を対象に、ペマフィブラート即放性製剤(IR錠)0.4mg/日又はプラセボを1日2回に分けて投与する無作為化プラセボ対照二重盲検比較試験を実施した。追跡期間は3.32年(中央値)であった。主要評価項目である心血管イベント(非致死性心筋梗塞、非致死性虚血性脳卒中、冠動脈血行再建術、心血管死のいずれか)の初回発現までの期間を評価した結果、複合エンドポイントの発現率(100人・年当たりのイベント発症例数)はIR錠群及びプラセボ群でそれぞれ3.53及び3.40で、ハザード比[95%信頼区間]は1.03[0.92,1.16]であった。副次評価項目である投与4ヵ月時におけるIR錠群の空腹時TGのベースライン(3.340±1.0157mmol/L(平均値±標準偏差)、n=5,224)からの変化率は−31.073%(中央値、n=4,814)、プラセボ群の空腹時TGのベースライン(3.303±0.9922mmol/L(平均値±標準偏差)、n=5,241)からの変化率は−6.882%(中央値、n=4,848)であり、IR錠群とプラセボ群との変化率の差は−24.4%[−27.1,−21.6](最小二乗平均値[95%信頼区間]、エンドポイントの欠測を多重補完法を用いて補完した、性別、心血管疾患の既往歴、ベースライン時のスタチン使用及びベースラインの測定値を共変量とした共分散分析、n=5,240)であった。
副作用発現割合はIR錠群及びプラセボ群でそれぞれ8.2%(433/5,264例)及び8.4%(441/5,274例)であり、IR錠群で発現した主な副作用は血中クレアチンホスホキナーゼ増加0.7%(37/5,264例)、筋肉痛0.5%(25/5,264例)であった。
本試験で認められた有害事象のうち、肺塞栓症の発現割合はIR錠群及びプラセボ群でそれぞれ0.7%(37/5,264例)及び0.3%(16/5,274例)、深部静脈血栓症の発現割合はIR錠群及びプラセボ群でそれぞれ0.7%(36/5,264例)及び0.2%(13/5,274例)であり、IR錠群で発現割合が高かったが、IR錠との関連性は全症例で否定された。なお、日本人集団における肺塞栓症の発現割合はIR錠群及びプラセボ群でそれぞれ0.6%(1/160例)及び0%(0/145例)、深部静脈血栓症の発現割合はIR錠群及びプラセボ群でそれぞれ0.6%(1/160例)及び0.7%(1/145例)であった12。
副作用発現割合はIR錠群及びプラセボ群でそれぞれ8.2%(433/5,264例)及び8.4%(441/5,274例)であり、IR錠群で発現した主な副作用は血中クレアチンホスホキナーゼ増加0.7%(37/5,264例)、筋肉痛0.5%(25/5,264例)であった。
本試験で認められた有害事象のうち、肺塞栓症の発現割合はIR錠群及びプラセボ群でそれぞれ0.7%(37/5,264例)及び0.3%(16/5,274例)、深部静脈血栓症の発現割合はIR錠群及びプラセボ群でそれぞれ0.7%(36/5,264例)及び0.2%(13/5,274例)であり、IR錠群で発現割合が高かったが、IR錠との関連性は全症例で否定された。なお、日本人集団における肺塞栓症の発現割合はIR錠群及びプラセボ群でそれぞれ0.6%(1/160例)及び0%(0/145例)、深部静脈血栓症の発現割合はIR錠群及びプラセボ群でそれぞれ0.6%(1/160例)及び0.7%(1/145例)であった12。
18. 薬効薬理
18.1 作用機序
18.1.1 PPARαに対する活性は、PPARγ及びPPARδに対する活性に比べ強く、PPARαに対する選択的な活性化作用を示した16)(in vitro)。
18.1.2 肝臓でのTGの合成を抑制した16)(ラット)。
18.1.3 TGの肝臓から血中への分泌速度を有意に低下させた16)(ラット)。
18.1.4 LPL活性を増加させた16)(ラット)。
18.1.5 LPL活性を負に制御する因子であるApoC-III及びAngiopoietin-Like Protein3の血漿中濃度を有意に低下させ、肝臓における遺伝子(Apoc3、Angptl3)の発現を抑制した。また、LPL活性を阻害する遊離脂肪酸のβ酸化に関わる遺伝子(Aco、Cpt1a)の発現を亢進させた16)(ラット)。
18.1.6 血漿TGクリアランスを亢進させた16)(ラット)。
18.1.7 TG濃度を低下させHDL-コレステロール濃度を増加させる蛋白であるFGF21の血漿中濃度を増加させた16)(ラット)。
18.2 血漿脂質低下作用
フルクトース負荷高TG血症ラットへのペマフィブラートの経口投与により、用量依存的に血漿TG濃度が低下した16)。
18.3 HDL-コレステロール増加作用
ヒトApoA-Iトランスジェニックマウスへのペマフィブラートの経口投与により、血漿HDL-コレステロール濃度及びヒトApoA-I濃度が増加した16)。
18.4 抗動脈硬化作用
高脂肪・高コレステロール食を負荷したLDL受容体欠損マウスへのペマフィブラートの経口投与により、大動脈洞の脂質沈着面積が減少した16)。
18.5 血中コレステロール低下作用
19. 有効成分に関する理化学的知見
19.1. ペマフィブラート
| 一般的名称 | ペマフィブラート |
|---|---|
| 一般的名称(欧名) | Pemafibrate |
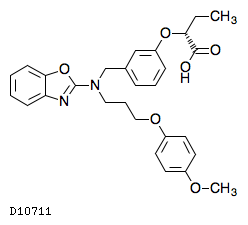
| 化学名 | (2R)-2-[3-({1,3-Benzoxazol-2-yl[3-(4-methoxyphenoxy)propyl]amino}methyl)phenoxy]butanoic acid |
| 分子式 | C28H30N2O6 |
| 分子量 | 490.55 |
| 融点 | 95〜101℃ |
| 物理化学的性状 | 白色の粉末である。ジメチルスルホキシド、N,N-ジメチルホルムアミドに溶けやすく、メタノールにやや溶けやすく、アセトニトリル、エタノール(99.5)にやや溶けにくく、水にほとんど溶けない。 |
| 分配係数 | (log P):4.63(pH2)、4.62(pH4)、2.87(pH6)、1.78(pH8)、1.59(pH10)、1.63(pH12) [1-オクタノール/Britton-Robinson緩衝液(20±1℃)] |
| KEGG DRUG |
|
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
<パルモディアXR錠0.2mg>
PTP
100錠(10錠×10)、500錠(10錠×50)
バラ
500錠(プラスチックボトル)
<パルモディアXR錠0.4mg>
PTP
100錠(10錠×10)、500錠(10錠×50)
バラ
500錠(プラスチックボトル)
23. 主要文献
- 興和(株)社内資料:徐放性製剤の第II相臨床薬理試験(2023年6月26日承認、CTD2.7.6.1)
- 興和(株)社内資料:第I相マスバランス試験(海外)(パルモディア錠0.1mg:2017年7月3日承認、CTD2.7.6.1)
- 興和(株)社内資料:非臨床試験 薬物動態試験(パルモディア錠0.1mg:2017年7月3日承認、CTD2.6.4.1-10)
- 興和(株)社内資料:第III相腎機能障害患者を対象とした薬物動態試験(パルモディア錠0.1mg:2017年7月3日承認、CTD2.7.6.6)
- 興和(株)社内資料:腎機能障害患者を対象とした製造販売後臨床試験
- 興和(株)社内資料:第III相肝機能障害患者を対象とした薬物動態試験(パルモディア錠0.1mg:2017年7月3日承認、CTD2.7.6.5)
- 興和(株)社内資料:薬物相互作用試験[1](海外)(パルモディア錠0.1mg:2017年7月3日承認、CTD2.7.6.13-17、2.7.6.29-30)
- 興和(株)社内資料:薬物相互作用試験[2](海外を含む)(パルモディア錠0.1mg:2017年7月3日承認、CTD2.7.6.8-11)
- 興和(株)社内資料:徐放性製剤の第III相検証試験(2023年6月26日承認、CTD2.7.6.3)
- 興和(株)社内資料:徐放性製剤の第III相長期投与試験(2023年6月26日承認、CTD2.7.6.4)
- 興和(株)社内資料:スタチン不耐の高コレステロール血症患者を対象とした徐放性製剤の第III相検証試験(2026年2月19日承認、CTD2.7.6.1)
- 興和(株)社内資料:製造販売後臨床試験(心血管アウトカム試験)
- Fruchart JC., Cardiovasc Diabetol., 12, 82, (2013) »PubMed
- Sahebkar A,et al., Expert Opin Pharmacother., 15, 493-503, (2014) »PubMed
- Pawlak M,et al., J Hepatol., 62, 720-33, (2015) »PubMed
- 興和(株)社内資料:非臨床試験 薬理試験(パルモディア錠0.1mg:2017年7月3日承認、CTD2.6.2.1-8)
- Takei K,et al., J Pharmacol Sci., 133, 214-22, (2017) »PubMed
- Tanigawa R,et al., J Atheroscler Thromb., 32, 823-39, (2025) »PubMed
24. 文献請求先及び問い合わせ先
文献請求先
興和株式会社
くすり相談センター 受付時間 9:00〜17:00(土・日・祝日・弊社休日を除く)
〒103-8433
東京都中央区日本橋本町三丁目4-14
電話:0120-508-514
03-3279-7587
03-3279-7587
製品情報問い合わせ先
興和株式会社
くすり相談センター 受付時間 9:00〜17:00(土・日・祝日・弊社休日を除く)
〒103-8433
東京都中央区日本橋本町三丁目4-14
電話:0120-508-514
03-3279-7587
03-3279-7587
26. 製造販売業者等
26.1 製造販売元
興和株式会社
東京都中央区日本橋本町三丁目4-14