医薬品情報
| 総称名 | リトゴビ |
|---|---|
| 一般名 | フチバチニブ |
| 欧文一般名 | Futibatinib |
| 製剤名 | フチバチニブ錠 |
| 薬効分類名 | 抗悪性腫瘍剤 FGFR注)阻害剤 注)FGFR:Fibroblast Growth Factor Receptor(線維芽細胞増殖因子受容体) |
| 薬効分類番号 | 4291 |
| ATCコード | L01EN04 |
| KEGG DRUG |
D11725
フチバチニブ
|
| JAPIC | 添付文書(PDF) |
添付文書情報2025年6月 改訂(第2版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| リトゴビ錠4mg | LYTGOBI tablets | 大鵬薬品工業 | 4291080F1029 | 10252.5円/錠 | 劇薬, 処方箋医薬品 |
1. 警告
本剤は、緊急時に十分対応できる医療施設において、がん化学療法に十分な知識・経験を持つ医師のもとで、本剤の使用が適切と判断される症例についてのみ投与すること。また、治療開始に先立ち、患者又はその家族に有効性及び危険性を十分説明し、同意を得てから投与すること。
2. 禁忌
次の患者には投与しないこと
本剤の成分に対し過敏症の既往歴のある患者
4. 効能または効果
がん化学療法後に増悪したFGFR2融合遺伝子陽性の治癒切除不能な胆道癌
5. 効能または効果に関連する注意
5.1 本剤の一次治療としての有効性及び安全性は確立していない。
5.2 本剤の術後補助療法における有効性及び安全性は確立していない。
5.3 臨床試験に組み入れられた患者の原発部位等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、適応患者の選択を行うこと。[17.1.1参照]
5.4 十分な経験を有する病理医又は検査施設における検査により、FGFR2融合遺伝子が確認された患者に投与すること。検査にあたっては、承認された体外診断用医薬品又は医療機器を用いること。なお、承認された体外診断用医薬品又は医療機器に関する情報については、以下のウェブサイトから入手可能である:
https://www.pmda.go.jp/review-services/drug-reviews/review-information/cd/0001.html
6. 用法及び用量
通常、成人には、フチバチニブとして1日1回20mgを空腹時に経口投与する。なお、患者の状態により適宜減量する。
7. 用法及び用量に関連する注意
7.1 他の抗悪性腫瘍剤との併用について、有効性及び安全性は確立していない。
7.2 食後に本剤を投与した場合、本剤のCmax及びAUCが低下するとの報告がある。食事の影響を避けるため、食事の1時間前から食後2時間までの間の服用は避けること。[16.2.1参照]
7.3 本剤投与により副作用が発現した場合には、以下の基準を考慮して休薬・減量・中止すること。[8.1、8.2、11.1.1、11.1.2参照]
| 減量レベル | 投与量 |
| 通常投与量 | 20mg |
| 1段階減量 | 16mg |
| 2段階減量 | 12mg |
| 3段階減量 | 投与中止 |
| 副作用 | 程度注) | 処置 |
| 網膜剥離 | − | ・症状がある場合又は検査で悪化が認められた場合は、本剤を休薬する。 ・休薬後、改善した場合は、1段階減量して本剤の投与を再開できる。改善しない場合は、本剤の投与を中止する。 |
| 高リン血症 | 血清リン濃度5.5mg/dL以上〜7mg/dL以下 | ・リン制限食に加え、高リン血症治療剤を投与する。 |
| 血清リン濃度7mg/dL超〜10mg/dL以下 | ・リン制限食に加え、1段階減量し、高リン血症治療剤を投与する。 ・1段階減量後2週間以内に血清リン濃度が7mg/dL以下に改善した場合は、1段階減量の用量で本剤の投与を継続できる。 ・1段階減量後2週間以内に血清リン濃度が7mg/dL以下に改善しない場合は、さらに1段階減量する。 ・2段階減量後2週間以内に血清リン濃度が7mg/dL以下に改善しない場合は、7mg/dL以下になるまで本剤を休薬する。休薬後7mg/dL以下に改善した場合は、休薬前の用量で本剤の投与を再開できる。 | |
| 血清リン濃度10mg/dL超 | ・リン制限食に加え、高リン血症治療剤を投与する。 ・血清リン濃度が7mg/dL以下になるまで本剤を休薬する。休薬後7mg/dL以下に改善した場合は、1段階減量して本剤の投与を再開できる。 ・2段階減量後、血清リン濃度が10mg/dLを超えた場合は、本剤の投与を中止する。 | |
| 上記以外の副作用 | Grade3 | ・Grade1以下又はベースラインに回復するまで本剤を休薬する。回復後、1段階減量して本剤の投与を再開できる。なお、血液毒性について、1週間以内に回復した場合は、同一用量で本剤の投与を再開できる。 |
| Grade4 | ・本剤の投与を中止する。 |
8. 重要な基本的注意
9. 特定の背景を有する患者に関する注意
9.3 肝機能障害患者
9.3.1 中等度以上の肝機能障害患者(Child-Pugh分類B又はC)
減量を考慮するとともに、患者の状態をより慎重に観察し、副作用の発現に十分注意すること。本剤の血中濃度が上昇することがあり、副作用が強くあらわれるおそれがある。[16.6.1参照]
9.4 生殖能を有する者
9.4.1 妊娠する可能性のある女性には、本剤投与中及び最終投与後一週間において避妊する必要性及び適切な避妊法について説明すること。[9.5参照]
9.4.2 男性には、本剤投与中及び最終投与後一週間においてバリア法(コンドーム)を用いて避妊する必要性について説明すること。[9.5参照]
9.5 妊婦
9.6 授乳婦
9.7 小児等
小児等を対象とした臨床試験は実施していない。
10. 相互作用
相互作用序文
薬物代謝酵素用語
CYP3A
薬物代謝酵素用語
10.2 併用注意
| 強い又は中程度のCYP3A誘導剤 リファンピシン、カルバマゼピン、エファビレンツ等 [16.7.1、16.7.2参照] | 本剤の有効性が減弱するおそれがあるので、これらの薬剤との併用は可能な限り避け、CYP3A誘導作用のない又は弱い薬剤への代替を考慮すること。 | これらの薬剤がCYP3Aを誘導することにより、本剤の血中濃度が低下する可能性がある。 |
| 強い又は中程度のCYP3A阻害剤 イトラコナゾール、クラリスロマイシン、フルコナゾール等 [16.7.3、16.7.4参照] | 本剤の副作用が増強されるおそれがあるので、これらの薬剤との併用は可能な限り避けること。やむを得ず併用する場合には、本剤の減量を考慮するとともに、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | これらの薬剤がCYP3Aを阻害することにより、本剤の血中濃度が上昇する可能性がある。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 網膜剥離
11.2 その他の副作用
| 20%以上 | 5〜20%未満 | 5%未満 | |
| 眼障害 | ドライアイ、睫毛の異常、霧視 | 眼瞼炎、視力障害 | |
| 消化器障害 | 口内乾燥(30.1%)、下痢、口内炎 | 便秘、悪心、嘔吐 | |
| 一般全身障害 | 疲労 | ||
| 肝胆道系障害 | AST増加、ALT増加 | ||
| 腎障害 | 血中クレアチニン増加 | ||
| 代謝、栄養障害 | 食欲不振 | 高カルシウム血症、低ナトリウム血症、脱水 | |
| 筋骨格系 | 筋肉痛、関節痛、筋痙縮、CK上昇 | 四肢痛 | |
| 神経障害 | 味覚異常 | 末梢性ニューロパチー | 浮動性めまい |
| 皮膚障害 | 爪の異常(46.6%)、脱毛症(33.0%)、皮膚乾燥、手掌・足底発赤知覚不全症候群 | そう痒症 | 発疹 |
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
15. その他の注意
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
日本人進行固形癌患者に本剤20mgを空腹時に単回経口投与したときのフチバチニブの血漿中濃度推移及び薬物動態パラメータは以下のとおりであった4)。
日本人進行固形癌患者に本剤20mgを単回経口投与したときのフチバチニブの血漿中濃度推移(平均値+標準偏差)
| 投与量 (症例数) | Cmax(ng/mL) | Tmax(h) | AUC0-24(h・ng/mL) |
| 20mg (n=7) | 253±161 | 2.00(1.00、3.95) | 981±716 |
16.1.2 反復投与
外国人進行固形癌患者に本剤20mgを1日1回反復経口投与したときのフチバチニブの蓄積率は、1.06〜1.27であった5)。
16.2 吸収
16.2.1 食事の影響
16.3 分布
フチバチニブのヒト血漿タンパク結合率は約95%であり、主にヒト血清アルブミン及びα1酸性糖タンパク質と結合した2)(in vitro)。
16.4 代謝
16.5 排泄
健康成人6例に14C標識体を含むフチバチニブ20mgの溶液を単回経口投与したとき、投与336時間までに投与した放射能の63.6%が糞中、6.47%が尿中に排泄された2)。尿中及び糞中に未変化体はほとんど排泄されなかった(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 肝機能障害患者
本剤20mgを単回投与したとき、肝機能正常被験者(16例)に対する[1]軽度(Child-Pugh分類A)の肝機能障害患者(8例)、[2]中等度(Child-Pugh分類B)の肝機能障害患者(8例)及び[3]重度(Child-Pugh分類C)の肝機能障害患者(6例)におけるフチバチニブのCmax及びAUCinfの幾何平均値の比は、それぞれ[1]1.14及び0.857、[2]1.18及び1.22、[3]1.18及び1.19であった。また、肝機能正常被験者(16例)に対する[4]軽度の肝機能障害患者(7例)、[5]中等度の肝機能障害患者(8例)及び[6]重度の肝機能障害患者(6例)の非結合形フチバチニブのCmax及びAUCinfの幾何平均値の比は、それぞれ[4]1.21及び0.937、[5]1.50及び1.56、[6]2.30及び2.32であった8)(外国人データ)。[9.3.1参照]
16.7 薬物相互作用
16.7.1 リファンピシン
16.7.2 カルバマゼピン、エファビレンツ
16.7.3 イトラコナゾール
16.7.4 クラリスロマイシン、フルコナゾール
16.7.5 その他
(1)ランソプラゾール
健康成人20例にランソプラゾール(プロトンポンプ阻害剤)60mgを1日1回5日間反復投与し、本剤20mgを単回経口投与したとき、本剤単独投与時に対するランソプラゾール併用投与時のフチバチニブのCmax及びAUCinfの幾何平均値の比は、それぞれ1.08及び1.05であった11)(外国人データ)。
(2)ミダゾラム
健康成人24例に本剤20mgを1日1回7日間反復投与し、ミダゾラム(CYP3Aの基質)2mgを単回経口投与したとき、ミダゾラム単独投与時に対する本剤併用投与時のミダゾラムのCmax及びAUCinfの幾何平均値の比は、それぞれ0.946及び0.911であった12)(外国人データ)。
(3)ジゴキシン、ロスバスタチン
健康成人20例に本剤20mgを1日1回7日間反復投与し、ジゴキシン(P-gpの基質)0.25mg及びロスバスタチン(BCRPの基質)10mgを単回併用投与したとき、ジゴキシン及びロスバスタチン単独投与時に対する本剤併用投与時のジゴキシンのCmax及びAUCinfの幾何平均値の比は、それぞれ0.951及び1.002、ロスバスタチンのCmax及びAUCinfの幾何平均値の比は、それぞれ1.102及び1.135であった13)(外国人データ)。
(4)キニジン
健康成人15例にキニジン(P-gp阻害剤)200mgを1日4回4日間反復投与し、本剤20mgを単回経口投与したとき、本剤単独投与時に対するキニジン併用投与時のフチバチニブのCmax及びAUCinfの幾何平均値の比は、それぞれ1.08及び1.17であった13)(外国人データ)。
(5)フチバチニブはCYP1A2の誘導作用を示した2)(in vitro)。
(6)フチバチニブはBCRPの基質である2)(in vitro)。
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国際共同第I/II相試験(TAS-120-101試験)
化学療法歴のあるFGFR2融合遺伝子陽性注)の治癒切除不能な肝内胆管癌を対象とした第II相パートにおいて、103例(うち日本人患者14例)に本剤20mgを1日1回経口投与した。主要評価項目であるRECIST ver.1.1に基づく独立評価判定による奏効率(%)は、41.7(95%信頼区間:32.1-51.9)であった14)。
注)FGFR2遺伝子のイントロン17とエクソン18の境界領域周辺に切断点を有し、他の遺伝子(リーディングフレームの一致の有無に関わらない)又は遺伝子間領域と融合したものと定義された。
本剤が投与された103例中102例(99.0%)に副作用が認められた。主な副作用は、高リン血症(85.4%)、脱毛症(33.0%)及び口内乾燥(30.1%)等であった。[5.3参照]
18. 薬効薬理
18.1 作用機序
フチバチニブは、線維芽細胞増殖因子受容体(FGFR)のチロシンキナーゼ活性を不可逆的に阻害する低分子化合物である。フチバチニブは、FGFR融合タンパク等のリン酸化を阻害し、下流のシグナル伝達分子のリン酸化を阻害することにより、腫瘍増殖抑制作用を示すと考えられている15)。
19. 有効成分に関する理化学的知見
19.1. フチバチニブ
| 一般的名称 | フチバチニブ |
|---|---|
| 一般的名称(欧名) | Futibatinib |
| 化学名 | 1-[(3S)-3-{4-Amino-3-[(3,5-dimethoxyphenyl)ethynyl]-1H-pyrazolo[3,4-d]pyrimidin-1-yl}pyrrolidin-1-yl]prop-2-en-1-one |
| 分子式 | C22H22N6O3 |
| 分子量 | 418.45 |
| 物理化学的性状 | 白色の結晶性の粉末である。 |
| KEGG DRUG |
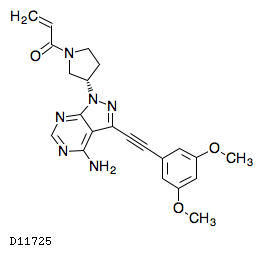
|
21. 承認条件
21.1 医薬品リスク管理計画を策定の上、適切に実施すること。
21.2 国内での治験症例が極めて限られていることから、製造販売後、一定数の症例に係るデータが集積されるまでの間は、全症例を対象に使用成績調査を実施することにより、本剤の使用患者の背景情報を把握するとともに、本剤の安全性及び有効性に関するデータを早期に収集し、本剤の適正使用に必要な措置を講じること。
22. 包装
PTP包装
35錠(5錠×7)
23. 主要文献
- 胚・胎児発生に関する試験(2023年6月26日承認、CTD 2.6.6.6)
- 薬物動態試験(2023年6月26日承認、CTD 2.7.2.1.2)
- 反復投与毒性試験(2023年6月26日承認、CTD 2.6.6.3)
- 臨床薬理試験(2023年6月26日承認、CTD 2.7.2.2.2.2)
- 臨床薬理試験(2023年6月26日承認、CTD 2.7.2.3.1.2)
- 臨床薬理試験(2023年6月26日承認、CTD 2.7.2.2.3.2)
- 薬物動態試験(2023年6月26日承認、CTD 2.7.2.2.3.4.2)
- 社内資料:薬物動態試験,研究報告書No.RR23PK001
- 臨床薬理試験(2023年6月26日承認、CTD 2.7.2.2.4.1)
- 臨床薬理試験(2023年6月26日承認、CTD 2.7.2.2.4.2)
- 臨床薬理試験(2023年6月26日承認、CTD 2.7.2.2.3.3)
- 臨床薬理試験(2023年6月26日承認、CTD 2.7.2.2.4.3)
- Long A,et al., Clin Transl Sci., 17 (9), e70012, (2024) »PubMed
- TAS-120-101試験第II相パート(2023年6月26日承認、CTD 2.7.6.9.2)
- Goyal,L,et al., Cancer Discov., 9 (8), 1064-1079, (2019) »PubMed
24. 文献請求先及び問い合わせ先
文献請求先
大鵬薬品工業株式会社
医薬品情報課
〒101-8444
東京都千代田区神田錦町1-27
電話:0120-20-4527
製品情報問い合わせ先
大鵬薬品工業株式会社
医薬品情報課
〒101-8444
東京都千代田区神田錦町1-27
電話:0120-20-4527
26. 製造販売業者等
26.1 製造販売元
大鵬薬品工業株式会社
東京都千代田区神田錦町1-27