医薬品情報
| 総称名 | ベンダムスチン塩酸塩 |
|---|---|
| 一般名 | ベンダムスチン塩酸塩水和物 |
| 欧文一般名 | Bendamustine Hydrochloride Hydrate |
| 製剤名 | ベンダムスチン塩酸塩水和物注射剤 |
| 薬効分類名 | 抗悪性腫瘍剤 |
| 薬効分類番号 | 4219 |
| ATCコード | L01AA09 |
| KEGG DRUG |
D11661
ベンダムスチン塩酸塩水和物
|
| KEGG DGROUP |
DG00678
ベンダムスチン
|
| JAPIC | 添付文書(PDF) |
添付文書情報2025年9月 改訂(第2版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ベンダムスチン塩酸塩点滴静注液25mg/1mL「イセイ」 (後発品) | Bendamustine Hydrochloride for I.V.Infusion 25mg/1mL"ISEI" | コーアイセイ | 4219405A2048 | 7134円/瓶 | 劇薬, 処方箋医薬品注) |
| ベンダムスチン塩酸塩点滴静注液100mg/4mL「イセイ」 (後発品) | Bendamustine Hydrochloride for I.V.Infusion 100mg/4mL"ISEI" | コーアイセイ | 4219405A1050 | 23450円/瓶 | 劇薬, 処方箋医薬品注) |
1. 警告
2. 禁忌
次の患者には投与しないこと
2.1 本剤の成分に対し重篤な過敏症の既往歴のある患者
2.2 妊婦又は妊娠している可能性のある女性[9.5参照]
4. 効能または効果
5. 効能または効果に関連する注意
<未治療の低悪性度B細胞性非ホジキンリンパ腫及びマントル細胞リンパ腫>
<再発又は難治性のびまん性大細胞型B細胞リンパ腫>
6. 用法及び用量
<低悪性度B細胞性非ホジキンリンパ腫>
○抗CD20抗体併用の場合
通常、成人には、ベンダムスチン塩酸塩として90mg/m2(体表面積)を1日1回10分又は1時間かけて点滴静注する。投与を2日間連日行い、26日間休薬する。これを1サイクルとして、投与を繰り返す。なお、患者の状態により適宜減量する。
○単独投与の場合(再発又は難治性の場合に限る)
通常、成人には、ベンダムスチン塩酸塩として120mg/m2(体表面積)を1日1回10分又は1時間かけて点滴静注する。投与を2日間連日行い、19日間休薬する。これを1サイクルとして、投与を繰り返す。なお、患者の状態により適宜減量する。
<マントル細胞リンパ腫>
○未治療の場合
リツキシマブ(遺伝子組換え)との併用において、通常、成人には、ベンダムスチン塩酸塩として90mg/m2(体表面積)を1日1回10分又は1時間かけて点滴静注する。投与を2日間連日行い、26日間休薬する。これを1サイクルとして、投与を繰り返す。なお、患者の状態により適宜減量する。
○再発又は難治性の場合
通常、成人には、ベンダムスチン塩酸塩として120mg/m2(体表面積)を1日1回10分又は1時間かけて点滴静注する。投与を2日間連日行い、19日間休薬する。これを1サイクルとして、投与を繰り返す。なお、患者の状態により適宜減量する。
<再発又は難治性のびまん性大細胞型B細胞リンパ腫>
○リツキシマブ(遺伝子組換え)併用の場合
通常、成人には、ベンダムスチン塩酸塩として120mg/m2(体表面積)を1日1回10分又は1時間かけて点滴静注する。投与を2日間連日行い、19日間休薬する。これを1サイクルとして、最大6サイクル投与を繰り返す。なお、患者の状態により適宜減量する。
○リツキシマブ(遺伝子組換え)及びポラツズマブ ベドチン(遺伝子組換え)併用の場合
通常、成人には、ベンダムスチン塩酸塩として90mg/m2(体表面積)を1日1回10分又は1時間かけて点滴静注する。投与を2日間連日行い、19日間休薬する。これを1サイクルとして、最大6サイクル投与を繰り返す。なお、患者の状態により適宜減量する。
<腫瘍特異的T細胞輸注療法の前処置>
再生医療等製品の用法及び用量又は使用方法に基づき使用する。
7. 用法及び用量に関連する注意
<効能共通>
7.1 本剤による治療中に高度の骨髄抑制が認められた場合には、次のような目安により、適切に休薬、減量又は投与中止を考慮すること。[11.1.1参照]
| 投与間隔又は投与量の調節 | 指標 | |
| 休薬 | 次サイクル投与開始にあたり、骨髄抑制が下記の指標に回復するまで休薬すること。 | |
| 低悪性度B細胞性非ホジキンリンパ腫、マントル細胞リンパ腫(リツキシマブ(遺伝子組換え)併用及び単独投与の場合)及び再発又は難治性のびまん性大細胞型B細胞リンパ腫の場合 | 好中球数1,000/mm3以上及び血小板数75,000/mm3以上 | |
| 未治療のマントル細胞リンパ腫の場合<アカラブルチニブマレイン酸塩水和物及びリツキシマブ(遺伝子組換え)併用の場合> | 好中球数1,000/mm3以上、血小板数50,000/mm3以上及びその他の血液毒性がGrade 2注1以下又はベースライン | |
| 減量又は中止 | 治療中に、下記の指標に該当する骨髄抑制が認められた場合には、休薬の項の指標に回復したことを確認の上、次サイクルの投与を開始すること。その場合、以下のとおり減量又は投与中止を考慮すること。 | |
| 低悪性度B細胞性非ホジキンリンパ腫及びマントル細胞リンパ腫(リツキシマブ(遺伝子組換え)併用及び単独投与の場合)の場合 | 好中球数500/mm3未満又は血小板数25,000/mm3未満 | |
| ・前サイクル投与量120mg/m2の場合:90mg/m2に減量 ・前サイクル投与量90mg/m2の場合:60mg/m2に減量 ・前サイクル投与量60mg/m2の場合:投与中止 なお、減量を行った場合には、以降投与量を維持し、増量しないこと。 | ||
| 再発又は難治性のびまん性大細胞型B細胞リンパ腫の場合 <リツキシマブ(遺伝子組換え)併用の場合> | 好中球数500/mm3未満、好中球数1,000/mm3未満が2週間以上持続する、又は血小板数75,000/mm3未満 | |
| ・前サイクル投与量120mg/m2の場合:90mg/m2に減量 ・前サイクル投与量90mg/m2の場合:60mg/m2に減量 ・前サイクル投与量60mg/m2の場合:投与中止 なお、減量を行った場合には、以降投与量を維持し、増量しないこと。 | ||
| 再発又は難治性のびまん性大細胞型B細胞リンパ腫の場合 <リツキシマブ(遺伝子組換え)及びポラツズマブ ベドチン(遺伝子組換え)併用の場合> | 好中球数1,000/mm3未満又は血小板数50,000/mm3未満 | |
| 次サイクル投与予定日の7日目までに休薬の項の指標に回復した場合は、減量せずに投与し、8日目以降に回復した場合は、以下のとおり減量又は投与を中止すること。 ・前サイクル投与量90mg/m2の場合:70mg/m2に減量 ・前サイクル投与量70mg/m2の場合:50mg/m2に減量 ・前サイクル投与量50mg/m2の場合:投与中止 なお、減量を行った場合には、以降投与量を維持し、増量しないこと。 | ||
| 未治療のマントル細胞リンパ腫の場合<アカラブルチニブマレイン酸塩水和物及びリツキシマブ(遺伝子組換え)併用の場合> | 好中球数1,000/mm3未満、血小板数50,000/mm3未満又はGrade 4注1のその他の血液毒性 | |
| ・前サイクル投与量90mg/m2の場合:70mg/m2に減量 ・前サイクル投与量70mg/m2の場合:投与中止 | ||
7.2 本剤による治療中に非血液毒性が認められた場合には、次のような目安により、適切に休薬、減量又は投与中止を考慮すること。
| 投与間隔又は投与量の調節 | 指標 | |
| 休薬 | 次サイクル投与開始にあたり、臨床検査値等が下記の指標に回復するまで休薬すること。 | |
| 低悪性度B細胞性非ホジキンリンパ腫、マントル細胞リンパ腫(リツキシマブ(遺伝子組換え)併用及び単独投与の場合)及び再発又は難治性のびまん性大細胞型B細胞リンパ腫の場合 | Grade 2注1以下の非血液毒性 総ビリルビン:2.0mg/dL未満 血清クレアチニン:2.0mg/dL未満 | |
| 未治療のマントル細胞リンパ腫の場合<アカラブルチニブマレイン酸塩水和物及びリツキシマブ(遺伝子組換え)併用の場合> | Grade 1注1又はベースライン | |
| 減量又は中止 | 治療中に、下記の指標に該当する副作用が認められた場合には、休薬の項の指標に回復したことを確認の上、次サイクルの投与を開始すること。その場合、以下のとおり減量又は投与中止を考慮すること。 | |
| 低悪性度B細胞性非ホジキンリンパ腫、マントル細胞リンパ腫(リツキシマブ(遺伝子組換え)併用及び単独投与の場合)及び再発又は難治性のびまん性大細胞型B細胞リンパ腫の場合 | Grade 3注1以上の非血液毒性 | |
| ・前サイクル投与量120mg/m2の場合:90mg/m2に減量 ・前サイクル投与量90mg/m2の場合:60mg/m2に減量 ・前サイクル投与量60mg/m2の場合:投与中止 なお、減量を行った場合には、以降投与量を維持し、増量しないこと。 | ||
| 未治療のマントル細胞リンパ腫の場合<アカラブルチニブマレイン酸塩水和物及びリツキシマブ(遺伝子組換え)併用の場合> | Grade 3注1以上の非血液毒性 | |
| ・前サイクル投与量90mg/m2の場合:70mg/m2に減量 ・前サイクル投与量70mg/m2の場合:投与中止 | ||
注1:NCI-CTCAE Version 4.0
<低悪性度B細胞性非ホジキンリンパ腫及び未治療のマントル細胞リンパ腫>
<再発又は難治性のマントル細胞リンパ腫>
7.4 他の抗悪性腫瘍剤との併用について、有効性及び安全性は確立していない。
8. 重要な基本的注意
8.2 リンパ球減少が高頻度にあらわれ、重症の免疫不全が増悪又は発現することがあるので、頻回に臨床検査(血液検査等)を行うなど、免疫不全の兆候について綿密な検査を行うこと。カンジダ等の真菌、サイトメガロウイルス等のウイルス、ニューモシスティス等による重症日和見感染に注意すること。
また、B型肝炎ウイルスの再活性化による肝炎があらわれることがあるので、本剤投与に先立って肝炎ウイルス感染の有無を確認し、本剤投与前に適切な処置を行うこと。[9.1.2、9.1.4、11.1.1、11.1.2参照]
また、B型肝炎ウイルスの再活性化による肝炎があらわれることがあるので、本剤投与に先立って肝炎ウイルス感染の有無を確認し、本剤投与前に適切な処置を行うこと。[9.1.2、9.1.4、11.1.1、11.1.2参照]
8.3 本剤による治療後、二次発がんが発生したとの報告があるので、本剤の投与終了後も経過を観察するなど十分に注意すること。
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 骨髄抑制のある患者
9.1.2 感染症を合併している患者
9.1.3 心疾患(心筋梗塞、重度の不整脈等)を合併する又は既往歴のある患者
心疾患を悪化させるおそれがある。
9.1.4 肝炎ウイルスの感染又は既往を有する患者
9.2 腎機能障害患者
副作用が強くあらわれるおそれがある。
9.3 肝機能障害患者
副作用が強くあらわれるおそれがある。
9.4 生殖能を有する者
9.4.1 妊娠する可能性のある女性患者には、投与期間中及び投与終了後3カ月間は適切な避妊法を用いるよう指導すること。[9.5参照]
9.4.3 生殖可能な年齢の患者に投与する必要がある場合には、性腺に対する影響を考慮すること。[15.2参照]
9.5 妊婦
9.6 授乳婦
授乳しないことが望ましい。本剤の乳汁移行については不明であるが、本剤は乳癌耐性蛋白(BCRP)の基質である可能性があるため、乳汁移行の可能性があり、乳児が乳汁を介して本剤を摂取した場合、乳児に重篤な副作用が発現するおそれがある。[15.2参照]
9.7 小児等
小児等を対象とした有効性及び安全性を指標とした国内臨床試験は実施していない。
9.8 高齢者
患者の状態を十分に観察しながら投与すること。一般に高齢者では生理機能が低下していることが多い。
10. 相互作用
10.2 併用注意
| 他の抗悪性腫瘍剤 | 骨髄抑制等の副作用が増強することがある。 | 骨髄抑制作用を増強する可能性がある。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 骨髄抑制
11.1.2 感染症
11.1.3 間質性肺疾患(頻度不明)
発熱、咳嗽、呼吸困難、胸部X線検査異常等が認められた場合には投与を中止し、適切な処置を行うこと。
11.1.4 腫瘍崩壊症候群(0.8%)
急性腎障害に至るおそれがあるので、体内水分量を適切に維持し、血液生化学検査(特に尿酸及びカリウム)を行うなど患者の状態を十分に観察すること。
11.1.5 重篤な皮膚症状(頻度不明)
中毒性表皮壊死融解症(Toxic Epidermal Necrolysis: TEN)、皮膚粘膜眼症候群(Stevens-Johnson症候群)があらわれることがあるので、観察を十分に行い、発熱、口腔粘膜の発疹、口内炎等の症状があらわれた場合には投与を中止し、適切な処置を行うこと。
11.1.6 ショック、アナフィラキシー(頻度不明)
11.2 その他の副作用
| 10%以上 | 10%未満 | 頻度不明 | |
| 血液 | 貧血、イムノグロブリン(IgA、IgM、IgG)低下 | 溶血性貧血、発熱性好中球減少症、播種性血管内凝固、汎血球減少、単球減少、白血球増加、好中球増加、好酸球増加、リンパ球増加、ヘモグロビン増加 | CD4/CD8比低下、CD4/CD8比上昇、ヘマトクリット減少、網状赤血球減少、無顆粒球症 |
| 心・血管障害 | 静脈炎 | 不整脈(房室ブロック、洞性頻脈、上室性期外収縮、心室性期外収縮等)、動悸、心筋梗塞、心血管障害、心障害、心嚢液貯留、心不全、左室機能不全、循環虚脱、パジェット・シュレッター症候群、血管障害(血管痛)、低血圧、高血圧、高血圧クリーゼ、ほてり、潮紅、静脈血栓症、心電図QT延長、心電図ST-T部分異常、心電図T波振幅減少 | 心肺不全、出血 |
| 眼 | 眼そう痒症、眼充血、眼瞼紅斑、強膜出血、角膜炎、閃輝暗点、流涙増加 | ||
| 消化器 | 便秘、下痢、悪心、嘔吐 | 口角口唇炎、口腔障害、口腔内潰瘍形成、口内炎、口内乾燥、舌障害、舌炎、食道痛、消化不良、おくび、胃炎、胃障害、胃食道逆流性疾患、胃不快感、腹痛、下腹部痛、腹部膨満、びらん性十二指腸炎、イレウス、痔核、肛門出血 | 潰瘍性食道炎、胃腸出血、消化管運動過剰 |
| 肝臓 | ALT上昇、AST上昇等の肝機能異常 | 胆汁うっ滞、胆石症、胆嚢ポリープ、肝毒性、γ-GTP上昇、血中ビリルビン低下、高ビリルビン血症 | |
| 代謝・栄養系 | 食欲不振、LDH上昇 | 高血糖、低比重リポ蛋白増加、脱水、高アミラーゼ血症、高カリウム血症、高クロール血症、高トリグリセリド血症、高尿酸血症、ALP上昇、総蛋白低下、低アルブミン血症、低カリウム血症、低カルシウム血症、低ナトリウム血症、低マグネシウム血症、低リン酸血症 | ALP低下、高カルシウム血症 |
| 筋骨格系 | 関節痛、筋骨格硬直、筋肉痛、頚部痛、骨痛、四肢痛、側腹部痛、背部痛 | ||
| 精神神経系 | 回転性めまい、体位性めまい、浮動性めまい、感覚障害、感覚鈍麻、錯感覚、味覚異常、知覚過敏、嗅覚錯誤、無感情、認知症、睡眠障害、不眠症、眠気、末梢性ニューロパチー、ラクナ梗塞、頭痛、頭部不快感 | 抗コリン作動性症候群、失声症、運動失調、脳炎、気分変化 | |
| 泌尿器 | 腎機能障害、腎結石症、腎不全、血尿、蛋白尿、頻尿、膀胱刺激症状、クレアチニン上昇、β2ミクログロブリン増加、BUN上昇 | BUN低下 | |
| 呼吸器 | 肺塞栓症、肺障害、肺浸潤、過敏性肺臓炎、呼吸不全、胸水、上気道の炎症、口腔咽頭痛、口腔咽頭不快感、湿性咳嗽、咳嗽、アレルギー性鼻炎、鼻出血、鼻漏、しゃっくり | 原発性異型肺炎、肺線維症、肺機能異常 | |
| 皮膚注1 | 発疹(23.0%) | 皮膚炎、ざ瘡様皮膚炎、アレルギー性皮膚炎、剥脱性皮膚炎、皮膚びらん、皮膚乳頭腫、皮膚剥脱、皮膚疼痛、乾皮症、乾癬、多形紅斑、紅斑、蕁麻疹、斑状丘疹状皮疹、湿疹、そう痒症、過敏性血管炎、脱毛症、手掌・足底発赤知覚不全症候群、色素沈着障害、寝汗、多汗症 | そう痒性皮疹、点状出血 |
| 注射部位 | 注射部位血管外漏出、注射部位反応(発赤、疼痛、硬結等) | ||
| その他 | 発熱、疲労、倦怠感 | C-反応性蛋白増加、浮腫、注入に伴う反応、過敏症、悪寒、熱感、口渇、低体温、粘膜の炎症、外耳の炎症、耳管閉塞、耳鳴、無力症、不規則月経、無月経、体重減少、体重増加、サルコイドーシス、胸痛、胸部不快感、疼痛、腫瘍疼痛、節足動物刺傷アレルギー、全身健康状態悪化 | 不妊症、尿中ウロビリン陽性、多臓器不全 |
14. 適用上の注意
14.1 薬剤調製時の注意
14.1.1 1日用量の調製方法
患者の体表面積から換算した投与量に対応する必要量を抜き取り、投与時間に応じて以下のとおり希釈すること。なお、調製時には、手袋を着用することが望ましい。
(1)10分かけて投与する場合は50mLの生理食塩液に加えること。
(2)1時間かけて投与する場合は、生理食塩液で最終投与液を250mLに調製すること。
14.1.2 本剤が体部に付着した場合
直ちに石鹸及び多量の水で十分に洗い、眼は水で洗浄すること。
14.2 薬剤投与時の注意
14.2.1 点滴静注に際し、投与液が血管外に漏れると、投与部位に紅斑、腫脹、疼痛、壊死を起こすことがあるので、投与液が血管外に漏れないように投与すること。血管外に漏れた場合は、速やかに投与を中止し、適切な処置を行うこと。
14.2.2 調製後は速やかに使用すること。なお、保存する必要がある場合には、室温保存では6時間以内、2〜8℃保存の場合は24時間以内に投与を終了すること。
15. その他の注意
15.2 非臨床試験に基づく情報
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与(1時間投与)
日本人患者に、ベンダムスチン塩酸塩90又は120mg/m2/日を1時間かけて点滴静注したときの薬物動態パラメータは以下のとおりであった1)。
| Dose(mg/m2) | 例数 | t1/2(hr) | Tmax(hr) | Cmax(ng/mL) | AUC0-t(ng・hr/mL) | Vz(mL) | CL(mL/hr) |
| 90 | 3 | 0.53±0.09 | 0.8±0.3 | 7250±3303 | 8327±3626 | 15075±4491 | 20246±8185 |
| 120 | 6 | 0.47±0.05 | 0.9±0.2 | 8616±4488 | 10212±5759 | 17532±10578 | 25963±15531 |
16.1.2 単回投与(リツキシマブ併用時、10分投与)
日本人患者に、リツキシマブ併用時にベンダムスチン塩酸塩90又は120mg/m2/日を10分かけて点滴静注したときの薬物動態パラメータは以下のとおりであった2)。
| Dose(mg/m2) | 例数 | t1/2(hr) | Cmax(ng/mL) | AUCinf(ng・hr/mL) | Vz(mL/kg) | CL(mL/hr/kg) |
| 90 | 6 | 0.43±0.11 | 9809±3413 | 4708±1732 | 353±161 | 565±185 |
| 120 | 6 | 0.50±0.07 | 16256±4434 | 8244±2796 | 327±92 | 464±156 |
16.1.3 10分投与と1時間投与の比較
外国人がん患者に、クロスオーバー法でベンダムスチン塩酸塩120mg/m2/日を10分かけて点滴静注したときと、120mg/m2/日を1時間かけて点滴静注したときの薬物動態パラメータは以下のとおりであった。ベンダムスチン塩酸塩の1時間投与時に対する10分投与時のAUC0-t及びAUCinfは、事前に規定された同等性の判断基準(幾何平均値の比の90%信頼区間が0.80〜1.25)の範囲内であった3)。
| 投与時間 | 例数 | t1/2(hr) | Cmax(ng/mL) | AUCinf(ng・hr/mL) | Vz(mL/kg) | CL(mL/hr/kg) |
| 10分 | 38 | 0.65±0.24 | 19158±6414 | 10370±5106 | 341±177 | 383±210 |
| 1時間 | 38 | 0.60±0.18 | 8868±4202 | 10528±5875 | 323±192 | 407±304 |
16.3 分布
ベンダムスチン塩酸塩のヒト血漿蛋白への結合率はin vitro試験で約94〜96%であり、α1酸性糖蛋白(<6%)よりもアルブミン(80〜92%)への結合率が高かった4)。
16.4 代謝
16.4.1 ヒト肝ミクロソームによるin vitro試験において、ベンダムスチン塩酸塩はCYP1A2によってgamma-hydroxybendamustine[M3]及びN-des-methylbendamustine[M4]に代謝され、また、非酵素的加水分解を受けることが確認された5)。
16.4.2 日本人患者にベンダムスチン塩酸塩120mg/m2/日を1時間かけて点滴静注したとき、M3及びM4の平均AUCは、M3で未変化体の6.3%、M4で1.2%であった1)。
16.5 排泄
16.5.1 日本人患者にベンダムスチン塩酸塩120mg/m2/日を1時間かけて点滴静注したとき、未変化体、M3及びM4の24時間尿中排泄率は、それぞれ投与量の1.6%、0.2%及び0.1%であった1)。
16.5.2 ラットに[14C]ベンダムスチンを静脈内投与後168時間までの尿・糞中放射能排泄率は尿中36.5%、糞中49.0%であり、イヌにおいては尿中22.2%、糞中66.4%であった8)。
16.6 特定の背景を有する患者
16.6.1 肝機能又は腎機能障害者における薬物動態
がん患者において、肝・腎機能正常の場合と肝機能障害(肝への浸潤・転移が30%〜70%)又は腎機能障害(クレアチニンクリアランスが60mL/min以下)がある場合を比較するために、ベンダムスチン塩酸塩120mg/m2/日を30分点滴静注後の薬物動態を評価した。肝・腎機能正常、肝機能障害及び腎機能障害者における薬物動態パラメータは以下のとおりであった(外国人データ)9)10)。
| 例数 | Tmax(min) | Cmax(ng/mL) | t1/2(min) | AUC0-t(hr・ng/mL) | |
| 肝・腎機能正常 | 12 | 29.6±7.2 | 10780±7024 | 28.2±15.9 | 11654±10590 |
| 肝機能障害 | 12 | 29.6±4.0 | 9893±3335 | 26.9±7.6 | 8868±4260 |
| 腎機能障害注1 | 12 | 31.3±10.0 | 9749±2542 | 26.4±6.4 | 8013±3404 |
17. 臨床成績
17.1 有効性及び安全性に関する試験
<再発又は難治性の低悪性度B細胞性非ホジキンリンパ腫及びマントル細胞リンパ腫>
17.1.1 国内第II相臨床試験(2007002試験)
がん化学療法又は抗体療法の治療歴を有する低悪性度B細胞性非ホジキンリンパ腫又はマントル細胞リンパ腫の患者を対象に、ベンダムスチン塩酸塩を単独で投与(ベンダムスチン塩酸塩120mg/m2をDay1及びDay2に静脈内投与し、その後19日間休薬する。これを1サイクルとして、最大6サイクルまで投与)した。なお、ベンダムスチン塩酸塩は1時間かけて投与された。
臨床成績は以下のとおりであった11)12)。
臨床成績は以下のとおりであった11)12)。
| 疾患名 | 例数 | 奏効率 (完全寛解+部分寛解/例数) | 完全寛解率 (完全寛解/例数) | 1年無増悪生存率 |
| 低悪性度B細胞性非ホジキンリンパ腫 | 58例 | 89.7% (52/58例) | 65.5% (38/58例) | 70.4% |
| マントル細胞リンパ腫 | 11例 | 100% (11/11例) | 72.7% (8/11例) | 90.0% |
また、安全性評価対象例69例中69例(100%)に副作用が認められた。主な副作用は、リンパ球数減少(98.6%)、白血球数減少(97.1%)、好中球数減少(89.9%)、悪心(84.1%)、CD4リンパ球減少(78.3%)、血小板数減少(75.4%)、赤血球数減少(68.1%)、ヘモグロビン減少(66.7%)、食欲減退(60.9%)、血中乳酸脱水素酵素増加(50.7%)、便秘(46.4%)、C-反応性蛋白増加(44.9%)、嘔吐(42.0%)、疲労(40.6%)、血中免疫グロブリンM減少(40.6%)、発疹(39.1%)、アスパラギン酸アミノトランスフェラーゼ増加(36.2%)、アラニンアミノトランスフェラーゼ増加(34.8%)、体重減少(34.8%)、発熱(31.9%)、静脈炎(30.4%)等であった。
<未治療の低悪性度B細胞性非ホジキンリンパ腫及びマントル細胞リンパ腫>
17.1.2 国内第II相臨床試験(2011002試験)
未治療の低悪性度B細胞性非ホジキンリンパ腫又は造血幹細胞移植の適応とならないマントル細胞リンパ腫の患者を対象に、本剤とリツキシマブを併用にて(4週間を1サイクルとして、本剤90mg/m2をDay1及びDay2に、リツキシマブ375mg/m2を第1サイクルはDay0、第2サイクル以降はDay1に点滴静脈内投与し、その後少なくとも26日間休薬する)、最大6サイクルまで投与した。なお、本剤は1時間かけて投与された。
完全寛解率は、低悪性度B細胞性非ホジキンリンパ腫67.8%(40/59例)及びマントル細胞リンパ腫70.0%(7/10例)であった13)。
また、安全性評価対象例69例中69例(100%)に副作用が認められた。主な副作用は白血球数減少(100%)、リンパ球数減少(97.1%)、好中球数減少(94.2%)、CD4リンパ球減少(92.8%)、悪心(66.7%)、便秘(62.3%)、血小板数減少(55.1%)、倦怠感(53.6%)、低γグロブリン血症(52.2%)、食欲不振(43.5%)、注入に伴う反応(40.6%)、発疹(39.1%)、貧血(34.8%)、静脈炎(34.8%)、AST上昇(31.9%)、LDH増加(30.4%)等であった。[5.1、7.3参照]
完全寛解率は、低悪性度B細胞性非ホジキンリンパ腫67.8%(40/59例)及びマントル細胞リンパ腫70.0%(7/10例)であった13)。
また、安全性評価対象例69例中69例(100%)に副作用が認められた。主な副作用は白血球数減少(100%)、リンパ球数減少(97.1%)、好中球数減少(94.2%)、CD4リンパ球減少(92.8%)、悪心(66.7%)、便秘(62.3%)、血小板数減少(55.1%)、倦怠感(53.6%)、低γグロブリン血症(52.2%)、食欲不振(43.5%)、注入に伴う反応(40.6%)、発疹(39.1%)、貧血(34.8%)、静脈炎(34.8%)、AST上昇(31.9%)、LDH増加(30.4%)等であった。[5.1、7.3参照]
17.1.3 海外第III相臨床試験(NHL1-2003試験)
未治療の低悪性度B細胞性非ホジキンリンパ腫又はマントル細胞リンパ腫の患者を対象とした無作為化非盲検群間比較試験の成績概要は以下のとおりであった。本剤とリツキシマブの併用注1とR-CHOP注2を比較した。なお、本剤は1時間かけて投与された。
主要評価項目とされた治験責任医師判定による無増悪生存期間(PFS)の最終解析時の成績は、R-CHOP群の31.3カ月(中央値)に対して、本剤群では61.4カ月(中央値)であった。ただし、治験実施計画書に事前に規定されていない解析計画に基づくものであるため、R-CHOP群に対する本剤群の優越性は検証されていない14)。
また、本剤が投与された安全性評価対象例267例中263例(98.5%)に副作用が認められた。主な副作用は、白血球数減少(81.6%)、顆粒球数減少(54.3%)、嘔吐(40.8%)、ヘモグロビン減少(36.0%)、血小板数減少(27.3%)、発疹(24.3%)、トランスアミナーゼ上昇(22.1%)、発熱(20.6%)等であった。[5.1、7.3参照]
主要評価項目とされた治験責任医師判定による無増悪生存期間(PFS)の最終解析時の成績は、R-CHOP群の31.3カ月(中央値)に対して、本剤群では61.4カ月(中央値)であった。ただし、治験実施計画書に事前に規定されていない解析計画に基づくものであるため、R-CHOP群に対する本剤群の優越性は検証されていない14)。
また、本剤が投与された安全性評価対象例267例中263例(98.5%)に副作用が認められた。主な副作用は、白血球数減少(81.6%)、顆粒球数減少(54.3%)、嘔吐(40.8%)、ヘモグロビン減少(36.0%)、血小板数減少(27.3%)、発疹(24.3%)、トランスアミナーゼ上昇(22.1%)、発熱(20.6%)等であった。[5.1、7.3参照]
| 本剤群注1 N=274 | R-CHOP群注2 N=275 | |
| PFS(医師判定)注3 中央値(月)(95%信頼区間) ハザード比(99%信頼区間) P値注4 | 61.4(45.3-NA) | 31.3(25.4-40.7) |
| 0.607(0.43-0.86) p<0.0001 | ||
| PFS(独立評価委員会判定)注3 中央値(月)(95%信頼区間) ハザード比(99%信頼区間) P値注4 | N=182注5 30.6(23.6-33.3) | N=171注5 23.3(16.5-26.0) |
| 0.735(0.5-1.08) p=0.0420 | ||
| 全生存期間 中央値(月)(95%信頼区間) ハザード比(95%信頼区間) P値注4 | NA(NA-NA) | NA(NA-NA) |
| 0.846(0.61-1.17) p=0.3101 | ||
注1:4週間を1サイクルとして、本剤1回90mg/m2をDay1及び2に静脈内投与、並びにリツキシマブ375mg/m2をDay1に静脈内投与。なお、第1サイクルはリツキシマブをDay0に投与した。
注2:3週間を1サイクルとして、リツキシマブ375mg/m2、シクロホスファミド750mg/m2、ドキソルビシン塩酸塩50mg/m2及びビンクリスチン硫酸塩1.4mg/m2(最大2mg)をDay1に静脈内投与、並びにプレドニゾン(国内未承認)100mgをDay1〜5に経口投与。なお、第1サイクルはリツキシマブをDay0に投与した。
注3:PFSの評価は第3サイクル及び治療終了後、並びに以後、必要時とされ、両群間で評価間隔は異なっていた。
注4:優越性検定でのP値
注5:独立評価委員会評価可能対象集団。なお、治験実施計画書に規定されていなかった独立評価委員会判定を事後的に実施したが、組入れから判定まで長期間が経過していたこと等から、評価に必要なすべての画像情報を入手できなかった。
<未治療の低悪性度B細胞性非ホジキンリンパ腫>
17.1.4 国際共同第III相臨床試験(GALLIUM試験)
未治療のCD20陽性の低悪性度B細胞性非ホジキンリンパ腫患者1,401例(日本人129例を含む)を対象とした非盲検無作為化比較試験注6の成績概要は以下のとおりであった。オビヌツズマブ(遺伝子組換え)と化学療法注7(CHOP注8、CVP注9又は本剤注10)との併用注11(オビヌツズマブ/化学療法群)とリツキシマブと化学療法との併用注12(対照群)を比較した。なお、本剤は1時間かけて投与された。
濾胞性リンパ腫患者注13(1,202例、日本人123例を含む)において、オビヌツズマブ/化学療法群では、対照群に比べ主要評価項目である治験責任医師判定によるPFSの有意な延長が認められ(ハザード比[95%信頼区間]:0.66[0.51〜0.85]、[層別Log-rank検定:P=0.0012(有意水準両側0.012)]、2016年1月31日データカットオフ)、中央値[95%信頼区間]はオビヌツズマブ/化学療法群では未達[推定不能]、対照群では未達[47.1カ月〜推定不能]であった。また、本剤が併用された濾胞性リンパ腫患者686例において、本剤/オビヌツズマブ群では、本剤/リツキシマブ群に比べ治験責任医師判定によるPFSのハザード比[95%信頼区間]は0.61[0.43〜0.86]であった15)。
また、本剤が投与された濾胞性リンパ腫患者の安全性評価対象例676例(日本人12例を含む)中578例(85.5%)に副作用が認められた。主な副作用は、悪心(48.7%)、疲労(30.8%)、好中球減少症(26.6%)、注入に伴う反応(14.8%)、嘔吐(14.1%)、便秘(11.1%)等であった。[5.1、7.3参照]
濾胞性リンパ腫患者注13(1,202例、日本人123例を含む)において、オビヌツズマブ/化学療法群では、対照群に比べ主要評価項目である治験責任医師判定によるPFSの有意な延長が認められ(ハザード比[95%信頼区間]:0.66[0.51〜0.85]、[層別Log-rank検定:P=0.0012(有意水準両側0.012)]、2016年1月31日データカットオフ)、中央値[95%信頼区間]はオビヌツズマブ/化学療法群では未達[推定不能]、対照群では未達[47.1カ月〜推定不能]であった。また、本剤が併用された濾胞性リンパ腫患者686例において、本剤/オビヌツズマブ群では、本剤/リツキシマブ群に比べ治験責任医師判定によるPFSのハザード比[95%信頼区間]は0.61[0.43〜0.86]であった15)。
また、本剤が投与された濾胞性リンパ腫患者の安全性評価対象例676例(日本人12例を含む)中578例(85.5%)に副作用が認められた。主な副作用は、悪心(48.7%)、疲労(30.8%)、好中球減少症(26.6%)、注入に伴う反応(14.8%)、嘔吐(14.1%)、便秘(11.1%)等であった。[5.1、7.3参照]
注6:導入療法期(最大8サイクル)と、導入療法期終了時に部分奏効以上の奏効が認められた患者を対象に、維持療法期が設定された。
注7:CHOP、CVP又は本剤のいずれかとの併用。
注8:21日間を1サイクルとして、シクロホスファミド750mg/m2、ドキソルビシン塩酸塩50mg/m2及びビンクリスチン硫酸塩1.4mg/m2(最大2mg)をDay1に静脈内投与、並びにプレドニゾロン/プレドニゾン(国内未承認)/メチルプレドニゾロン80又は100mgをDay1〜5に経口投与。
注9:21日間を1サイクルとして、シクロホスファミド750mg/m2及びビンクリスチン硫酸塩1.4mg/m2(最大2mg)をDay1に静脈内投与、並びにプレドニゾロン/プレドニゾン(国内未承認)/メチルプレドニゾロン80又は100mgをDay1〜5に経口投与。
注10:28日間を1サイクルとして、本剤90mg/m2をDay1及びDay2に静脈内投与、並びに第1サイクルのDay1にプレドニゾロン/プレドニゾン(国内未承認)/メチルプレドニゾロン80又は100mgを経口又は静脈内投与。
注11:CHOP、CVP又は本剤との併用で、オビヌツズマブ1日1回1000mgを第1サイクルはDay1、8及びDay15、第2サイクル以降はDay1に静脈内投与した。維持療法期では、オビヌツズマブ1日1回1000mgを2カ月間間隔で最長2年間静脈内投与した。
注12:CHOP、CVP又は本剤との併用で、リツキシマブ1回375mg/m2を各サイクルのDay1に静脈内投与した。維持療法期では、リツキシマブ1回375mg/m2を2カ月間間隔で最長2年間静脈内投与した。
注13:オビヌツズマブの承認効能・効果は、CD20陽性の濾胞性リンパ腫である。
<再発又は難治性の低悪性度B細胞性非ホジキンリンパ腫>
17.1.5 海外第III相臨床試験(GADOLIN試験)
リツキシマブ治療抵抗性注14のCD20陽性の低悪性度B細胞性非ホジキンリンパ腫患者413例を対象とした非盲検無作為化比較試験注15の成績概要は以下のとおりであった。本剤とオビヌツズマブとの併用注16(本剤/オビヌツズマブ併用群)と本剤単独投与注17(対照群)を比較した注13。なお、本剤は1時間かけて投与された。
低悪性度B細胞性非ホジキンリンパ腫患者396例において、本剤/オビヌツズマブ併用群では、対照群に比べ主要評価項目である中央判定によるPFSの有意な延長が認められ(ハザード比[95%信頼区間]:0.55[0.40〜0.74]、[層別Log-rank検定:P=0.0001(有意水準両側0.015)]、2014年9月1日データカットオフ)、中央値[95%信頼区間]は本剤/オビヌツズマブ併用群では未達[22.5カ月〜推定不能]、対照群では14.9カ月[12.8〜16.6カ月]であった。また、濾胞性リンパ腫患者注13321例において、本剤/オビヌツズマブ併用群では、対照群に比べ中央判定によるPFSのハザード比[95%信頼区間]は0.48[0.34〜0.68]であった16)。
また、本剤が投与された濾胞性リンパ腫患者の安全性評価対象例330例中304例(92.1%)に副作用が認められた。主な副作用は、注入に伴う反応(55.5%)、悪心(52.4%)、疲労(31.8%)、好中球減少症(25.8%)等であった。[7.3参照]
低悪性度B細胞性非ホジキンリンパ腫患者396例において、本剤/オビヌツズマブ併用群では、対照群に比べ主要評価項目である中央判定によるPFSの有意な延長が認められ(ハザード比[95%信頼区間]:0.55[0.40〜0.74]、[層別Log-rank検定:P=0.0001(有意水準両側0.015)]、2014年9月1日データカットオフ)、中央値[95%信頼区間]は本剤/オビヌツズマブ併用群では未達[22.5カ月〜推定不能]、対照群では14.9カ月[12.8〜16.6カ月]であった。また、濾胞性リンパ腫患者注13321例において、本剤/オビヌツズマブ併用群では、対照群に比べ中央判定によるPFSのハザード比[95%信頼区間]は0.48[0.34〜0.68]であった16)。
また、本剤が投与された濾胞性リンパ腫患者の安全性評価対象例330例中304例(92.1%)に副作用が認められた。主な副作用は、注入に伴う反応(55.5%)、悪心(52.4%)、疲労(31.8%)、好中球減少症(25.8%)等であった。[7.3参照]
注14:リツキシマブを含む治療法に対して治療抵抗性の患者(直近のリツキシマブ療法(単剤療法か化学療法との併用のいずれか)に対して不応、又は治療終了後6カ月以内に病勢の進行が認められた患者)が対象とされた。
注15:導入療法期(最大6サイクル)と、導入療法期終了時に病勢進行が認められなかった患者を対象に、維持療法期が設定された。
注16:導入療法期では、28日間を1サイクルとし、第1サイクルではDay1、8及び15、第2〜6サイクルではDay1にオビヌツズマブ1回1000mgを静脈内投与、各サイクルのDay1及びDay2に本剤1回90mg/m2を静脈内投与し、最大6サイクル繰り返した。また、第1サイクルのDay1にプレドニゾロン/プレドニゾン(国内未承認)/メチルプレドニゾロン80又は100mgを経口又は静脈内投与した。維持療法期では、オビヌツズマブ1000mgを2カ月間間隔で最長2年間静脈内投与した。
注17:導入療法期では、28日間を1サイクルとし、各サイクルのDay1及びDay2に本剤1回120mg/m2を静脈内投与し、最大6サイクル繰り返した。維持療法期では、経過観察とされた。なお、本邦の承認用法・用量は、「21日間を1サイクルとし、各サイクルのDay1及びDay2に本剤1回120mg/m2を静脈内投与する」である。
<再発又は難治性のびまん性大細胞型B細胞リンパ腫>
17.1.6 国内第III相臨床試験(2017002試験)
再発又は難治性のびまん性大細胞型B細胞リンパ腫患者注18を対象に、本剤とリツキシマブを併用注19投与した。なお、本剤は1時間かけて投与された。
主要評価項目である奏効率は76.3%(29/38例、95%信頼区間:59.8〜88.6%)であった17)。
また、安全性評価対象例38例中37例(97.4%)に副作用が認められた。主な副作用は、リンパ球数減少(89.5%)、好中球数減少(81.6%)、白血球数減少(81.6%)、CD4リンパ球減少(65.8%)、血小板数減少(60.5%)等であった。[5.2参照]
主要評価項目である奏効率は76.3%(29/38例、95%信頼区間:59.8〜88.6%)であった17)。
また、安全性評価対象例38例中37例(97.4%)に副作用が認められた。主な副作用は、リンパ球数減少(89.5%)、好中球数減少(81.6%)、白血球数減少(81.6%)、CD4リンパ球減少(65.8%)、血小板数減少(60.5%)等であった。[5.2参照]
注18:[1]前治療数が2、かつ救援化学療法及び自家造血幹細胞移植が実施された、[2]前治療数が2、かつ自家造血幹細胞移植の適応とならず救援化学療法のみによる治療が実施された、又は[3]前治療数が1、かつ加齢、臓器機能低下等の理由により、2剤以上の抗悪性腫瘍剤の併用による救援化学療法の実施が困難と判断された、のいずれかを満たす患者が対象とされた。
注19:3週間を1サイクルとして、本剤1回120mg/m2をDay2及びDay3、及びリツキシマブ375mg/m2をDay1にそれぞれ静脈内投与した。最大6サイクル投与した。
17.1.7 海外第Ib/II相臨床試験(GO29365試験)
GO29365試験の第II相ランダム化パートにおいて、自家造血幹細胞移植の適応とならない再発又は難治性のびまん性大細胞型B細胞リンパ腫患者を対象とした非盲検無作為化比較試験の成績概要は以下のとおりであった。本剤、リツキシマブ及びポラツズマブ ベドチンとの併用注20(BR+Pola;40例)と本剤とリツキシマブの併用注20(BR;40例)を比較した。なお、本剤は1時間かけて投与された。
主要評価項目とされた独立評価委員会評価によるPrimary Response Assessment(PRA、本剤最終投与後6〜8週)時点におけるPET-CTを用いた完全奏効割合は、BR+Pola群では40.0%(16/40例)(95%信頼区間:24.9〜56.7%)、BR群では17.5%(7/40例)(95%信頼区間:7.3〜32.8%)であった(2018年4月30日データカットオフ)18)。
また、安全性評価対象例(BR+Pola群)39例中36例(92.3%)に副作用が認められた。主な副作用は、好中球減少症53.8%、血小板減少症41.0%、下痢及び貧血が各33.3%、疲労及び悪心が各23.1%、発熱及び末梢性ニューロパチーが各20.5%であった。[5.2参照]
主要評価項目とされた独立評価委員会評価によるPrimary Response Assessment(PRA、本剤最終投与後6〜8週)時点におけるPET-CTを用いた完全奏効割合は、BR+Pola群では40.0%(16/40例)(95%信頼区間:24.9〜56.7%)、BR群では17.5%(7/40例)(95%信頼区間:7.3〜32.8%)であった(2018年4月30日データカットオフ)18)。
また、安全性評価対象例(BR+Pola群)39例中36例(92.3%)に副作用が認められた。主な副作用は、好中球減少症53.8%、血小板減少症41.0%、下痢及び貧血が各33.3%、疲労及び悪心が各23.1%、発熱及び末梢性ニューロパチーが各20.5%であった。[5.2参照]
注20:3週間を1サイクルとし、各薬剤を最大6サイクル投与する。
・本剤(90mg/m2):第1サイクルではDay2及びDay3、第2〜6サイクルではDay1及びDay2
・リツキシマブ(375mg/m2):各サイクルDay1
・ポラツズマブ ベドチン(1.8mg/kg):第1サイクルDay2、第2〜6サイクルは各サイクルDay1
17.1.8 国内第II相臨床試験(JO40762試験)
自家造血幹細胞移植の適応とならない再発又は難治性のびまん性大細胞型B細胞リンパ腫患者を対象に、BR+Pola注20を投与した。なお、本剤は1時間かけて投与された。
主要評価項目である治験責任医師評価によるPRA時点におけるPET-CTを用いた完全奏効割合は34.3%(12/35例、95%信頼区間:19.1〜52.2%)であった(2019年12月24日データカットオフ)。また、安全性評価対象例35例中33例(94.3%)に副作用が認められた。主な副作用は、貧血37.1%、悪心31.4%、血小板減少症及び好中球減少症が各25.7%、便秘、血小板数減少及び好中球数減少が各22.9%、倦怠感及び食欲減退が各20.0%であった。[5.2参照]
主要評価項目である治験責任医師評価によるPRA時点におけるPET-CTを用いた完全奏効割合は34.3%(12/35例、95%信頼区間:19.1〜52.2%)であった(2019年12月24日データカットオフ)。また、安全性評価対象例35例中33例(94.3%)に副作用が認められた。主な副作用は、貧血37.1%、悪心31.4%、血小板減少症及び好中球減少症が各25.7%、便秘、血小板数減少及び好中球数減少が各22.9%、倦怠感及び食欲減退が各20.0%であった。[5.2参照]
<未治療の低悪性度B細胞性非ホジキンリンパ腫及びマントル細胞リンパ腫、再発又は難治性のびまん性大細胞型B細胞リンパ腫>
17.1.9 国内第I/II相臨床試験(2018001試験)
未治療の低悪性度B細胞性非ホジキンリンパ腫及びマントル細胞リンパ腫患者(グループ1)並びに再発又は難治性のびまん性大細胞型B細胞リンパ腫患者(グループ2)を対象に、本剤とリツキシマブを併用注21,22投与した。なお、本剤は、10分かけて投与された。有効性評価項目である奏効率は、グループ1及び2でそれぞれ93.1%(27/29例、95%信頼区間:77.2〜99.2%)及び66.7%(4/6例、95%信頼区間:22.3〜95.7%)であった2)。
また、グループ1の安全性評価対象例30例中30例(100%)に副作用が認められた。主な副作用は、リンパ球数減少(86.7%)、好中球数減少(83.3%)、白血球数減少(83.3%)、CD4リンパ球減少(73.3%)、悪心(73.3%)、血小板数減少(46.7%)、便秘(46.7%)、倦怠感(43.3%)等であった。また、グループ2の安全性評価対象例6例中6例(100%)に副作用が認められた。主な副作用は、リンパ球数減少(100%)、白血球数減少(100%)、悪心(83.3%)、好中球数減少(83.3%)、血小板数減少(83.3%)、倦怠感(66.7%)、食欲減退(66.7%)、貧血(50.0%)、CD4リンパ球減少(50.0%)等であった。[5.1、5.2、7.3参照]
また、グループ1の安全性評価対象例30例中30例(100%)に副作用が認められた。主な副作用は、リンパ球数減少(86.7%)、好中球数減少(83.3%)、白血球数減少(83.3%)、CD4リンパ球減少(73.3%)、悪心(73.3%)、血小板数減少(46.7%)、便秘(46.7%)、倦怠感(43.3%)等であった。また、グループ2の安全性評価対象例6例中6例(100%)に副作用が認められた。主な副作用は、リンパ球数減少(100%)、白血球数減少(100%)、悪心(83.3%)、好中球数減少(83.3%)、血小板数減少(83.3%)、倦怠感(66.7%)、食欲減退(66.7%)、貧血(50.0%)、CD4リンパ球減少(50.0%)等であった。[5.1、5.2、7.3参照]
注21:未治療の低悪性度B細胞性非ホジキンリンパ腫及びマントル細胞リンパ腫患者には、4週間を1サイクルとして、本剤90mg/m2をDay1及びDay2に静脈内投与、リツキシマブ375mg/m2を第1サイクルはDay0(第2サイクル以降はDay1)に静脈内投与し、最大6サイクルまで投与した。
注22:再発又は難治性のびまん性大細胞型B細胞リンパ腫患者には、3週間を1サイクルとして、本剤120mg/m2をDay2及びDay3に静脈内投与、リツキシマブ375mg/m2をDay1に静脈内投与し、最大6サイクルまで投与した。
18. 薬効薬理
18.1 作用機序
18.2 抗腫瘍作用
19. 有効成分に関する理化学的知見
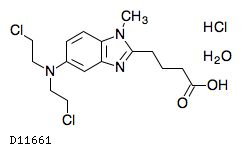
19.1. ベンダムスチン塩酸塩水和物
| 一般的名称 | ベンダムスチン塩酸塩水和物 |
|---|---|
| 一般的名称(欧名) | Bendamustine Hydrochloride Hydrate |
| 化学名 | 4-{5-[Bis(2-chloroethyl)amino]-1-methyl-1H-benzimidazol-2-yl}butanoic acid monohydrochloride monohydrate |
| 分子式 | C16H21Cl2N3O2・HCl・H2O |
| 分子量 | 412.74 |
| 物理化学的性状 | 本品は白色〜灰白色の粉末である。本品はメタノールに溶けやすく、エタノール(99.5)にやや溶けやすく、水にやや溶けにくい。 |
| KEGG DRUG |
|
20. 取扱い上の注意
包装開封後もバイアルを箱に入れて保存すること。
22. 包装
<ベンダムスチン塩酸塩点滴静注液25mg/1mL「イセイ」>
1包装あたり1バイアル(ガラスバイアル)
<ベンダムスチン塩酸塩点滴静注液100mg/4mL「イセイ」>
1包装あたり1バイアル(ガラスバイアル)
23. 主要文献
- 薬物動態(国内第I相臨床試験)(トレアキシン点滴静注用:2010年10月27日承認,申請資料概要2.7.6.1)
- 国内第I/II相臨床試験(18001試験)(トレアキシン点滴静注液:2022年2月25日承認,審査報告書)
- 海外第I相臨床試験(1301試験)(トレアキシン点滴静注液:2022年2月25日承認,審査報告書)
- 薬物動態[海外非臨床試験(KLG/06試験)](トレアキシン点滴静注用:2010年10月27日承認,申請資料概要2.6.4.4.5)
- 薬物動態(海外非臨床試験)(トレアキシン点滴静注用:2010年10月27日承認,申請資料概要2.6.4.5.5)
- Teichert J.,et al., Drug Metab.Dispos., 33, 984-992, (2005) »PubMed
- Teichert J.,et al., Drug Metab.Dispos., 37, 292-301, (2009) »PubMed
- 排泄(トレアキシン点滴静注用:2010年10月27日承認,申請資料概要2.6.4.6)
- 薬物動態[海外臨床試験(98B03試験)](トレアキシン点滴静注用:2010年10月27日承認,申請資料概要2.7.6.8)
- 肝機能障害又は腎機能障害患者を対象とした海外第I相試験(98B03試験)(トレアキシン点滴静注用:2010年10月27日承認,審査報告書)
- Ohmachi K.,et al., Cancer Sci., 101, 2059-2064, (2010) »PubMed
- 国内第II相臨床試験(2007002試験)(トレアキシン点滴静注用:2016年12月19日承認,申請資料概要2.7.6.8)
- Ogura M.,et al., Int.J.Hematol., 105, 470-477, (2017) »PubMed
- 海外第III相臨床試験(NHL 1-2003試験)(トレアキシン点滴静注用:2016年12月19日承認,申請資料概要2.7.6.1)
- Marcus R.,et al., N.Engl.J.Med., 377, 1331-1334, (2017) »PubMed
- Sehn LH.,et al., Lancet Oncol., 17, 1081-1093, (2016) »PubMed
- 国内第III相臨床試験(2017002試験)(トレアキシン点滴静注用:2021年3月23日承認,審査報告書)
- Sehn LH.,et al., J.Clin.Oncol., 38, 155-165, (2020) »PubMed
- Strumberg D.,et al., Anticancer Drugs, 7, 415-421, (1996) »PubMed
- Leoni L.M.,et al., Clin.Cancer Res., 14, 309-317, (2008) »PubMed
- Gaul L.,et al., J.Cancer Res.Clin.Oncol., 134, 245-253, (2008) »PubMed
- Roue G.,et al., Clin.Cancer Res., 14, 6907-6915, (2008) »PubMed
- Alonso R.,et al., Blood, 114, 1563-1575, (2009) »PubMed
- 薬効薬理[ヒト低悪性度B細胞性非ホジキンリンパ腫由来細胞株及びマントル細胞リンパ腫由来細胞株に対する細胞増殖抑制作用](トレアキシン点滴静注用:2010年10月27日承認,申請資料概要2.6.2.2)
- 悪性腫瘍由来細胞株に対する増殖抑制作用(トレアキシン点滴静注用:2021年3月23日承認,審査報告書)
24. 文献請求先及び問い合わせ先
文献請求先
高田製薬株式会社
文献請求窓口
〒336-8666
さいたま市南区沼影1丁目11番1号
電話:0120-989-813
FAX:048-838-2121
製品情報問い合わせ先
高田製薬株式会社
文献請求窓口
〒336-8666
さいたま市南区沼影1丁目11番1号
電話:0120-989-813
FAX:048-838-2121
26. 製造販売業者等
26.1 製造販売元
コーアイセイ株式会社
山形市若葉町13番45号
26.2 販売元
高田製薬株式会社
さいたま市西区宮前町203番地1