医薬品情報
| 総称名 | ハイイータン |
|---|---|
| 一般名 | グマロンチニブ水和物 |
| 欧文一般名 | Gumarontinib Hydrate |
| 製剤名 | グマロンチニブ水和物錠 |
| 薬効分類名 | 抗悪性腫瘍剤 MET阻害剤 |
| 薬効分類番号 | 4291 |
| KEGG DRUG |
D12705
グマロンチニブ水和物
|
| JAPIC | 添付文書(PDF) |
添付文書情報2024年9月 改訂(第2版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ハイイータン錠50mg | HAIYITAN tablets | 海和製薬 | 4291085F1021 | 4356.6円/錠 | 劇薬, 処方箋医薬品 |
1. 警告
2. 禁忌
次の患者には投与しないこと
本剤の成分に対し過敏症の既往歴のある患者
4. 効能または効果
MET遺伝子エクソン14スキッピング変異陽性の切除不能な進行・再発の非小細胞肺癌
5. 効能または効果に関連する注意
5.1 十分な経験を有する病理医又は検査施設における検査により、MET遺伝子エクソン14スキッピング変異が確認された患者に投与すること。検査にあたっては、承認された体外診断用医薬品又は医療機器を用いること。なお、承認された体外診断用医薬品又は医療機器に関する情報については、以下のウェブサイトから入手可能である:
https://www.pmda.go.jp/review-services/drug-reviews/review-information/cd/0001.html
5.2 本剤の術前・術後補助療法における有効性及び安全性は確立していない。
6. 用法及び用量
通常、成人にはグマロンチニブとして1回300mgを1日1回空腹時に経口投与する。なお、患者の状態により適宜減量する。
7. 用法及び用量に関連する注意
7.1 他の抗悪性腫瘍剤との併用について、有効性及び安全性は確立していない。
7.2 食後に本剤を投与した場合、本剤のCmax及びAUCが上昇するとの報告がある。食事の影響を避けるため、食事の1時間前から食後2時間までの間の服用は避けること。[16.2.1参照]
7.3 副作用が発現した場合は、以下の基準を考慮して、本剤を休薬、減量又は中止すること。
| 減量レベル | 1回投与量 |
| 通常投与量 | 300mg |
| 1段階減量 | 250mg |
| 2段階減量 | 200mg |
| 3段階減量 | 150mg |
| 中止 | 150mgで忍容不能な場合、投与を中止する。 |
| 副作用 | 程度注) | 処置 |
| 間質性肺疾患 | Grade 1以上 | 投与を中止する。 |
| 肝機能障害 | Grade 2の血中ビリルビン増加 | Grade 1以下又はベースラインに回復するまで休薬する。 7日以内に回復した場合は、同一用量で投与を再開できる。 7日を過ぎてから回復した場合は、1段階減量して投与を再開できる。 |
| Grade 3の血中ビリルビン増加、AST又はALT増加 | Grade 1以下又はベースラインに回復するまで休薬する。回復後、1段階減量して投与を再開できる。 | |
| Grade 4 | 投与を中止する。 | |
| 浮腫 | Grade 2かつ対症療法により回復しない場合 | Grade 1以下に回復するまで休薬する。 3〜5日以内に回復した場合は、同一用量で投与を再開できる。同一用量で再開後に再度Grade 2に悪化した場合は、1段階減量する。 3〜5日を過ぎてから回復した場合は、1段階減量して投与を再開できる。 |
| Grade 3 | Grade 2以下に回復するまで休薬し、1段階減量して投与を再開できる。 | |
| Grade 4 | 投与を中止する。 | |
| 血液障害 | Grade 3又は4 | Grade 1以下に回復するまで休薬し、1段階減量して投与を再開できる。 |
| 上記以外の副作用 | Grade 3 | Grade 1以下に回復するまで休薬し、1段階減量して投与を再開できる。 |
| Grade 4 | 投与を中止する。 |
8. 重要な基本的注意
8.1 間質性肺疾患があらわれることがあるので、初期症状(息切れ、呼吸困難、咳嗽、発熱等)の確認及び定期的な胸部画像検査の実施等、観察を十分に行うこと。また、患者に対して、初期症状があらわれた場合には、速やかに医療機関を受診するよう指導すること。[1.2、9.1.1、11.1.1参照]
8.2 肝機能障害があらわれることがあるので、本剤投与開始前及び投与中は定期的に肝機能検査を行い、患者の状態を十分に観察すること。[11.1.3参照]
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 間質性肺疾患のある患者又はその既往歴のある患者
9.1.2 QT間隔延長のおそれ又はその既往歴のある患者
9.3 肝機能障害患者
本剤は主に代謝により体内から消失するため、血中濃度が上昇する可能性がある。なお、重度(総ビリルビンが基準値上限の3倍超)の肝機能障害患者を対象とした臨床試験は実施していない。[16.6.1参照]
9.4 生殖能を有する者
妊娠する可能性のある女性には、本剤投与中及び最終投与後1週間において避妊する必要性及び適切な避妊法について説明すること。[9.5参照]
9.5 妊婦
9.6 授乳婦
授乳しないことが望ましい。乳汁移行に関するデータはないが、乳児が乳汁を介して本剤を摂取した場合、乳児に重大な副作用が発現するおそれがある。[9.5参照]
9.7 小児等
小児等を対象とした臨床試験は実施していない。
10. 相互作用
相互作用序文
本剤は、多剤及び毒素排出タンパク質1(MATE1)並びにMATE2-Kに対する阻害作用を示す。
薬物代謝酵素用語
多剤及び毒素排出タンパク質1(MATE1)
薬物代謝酵素用語
多剤及び毒素排出タンパク質 MATE2-K
10.2 併用注意
| MATE1及びMATE2-Kの基質となる薬剤 メトホルミン等 [16.7.1参照] | これらの薬剤の副作用が増強されるおそれがあるため、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | 本剤がMATE1及びMATE2-Kを阻害することにより、これらの薬剤の血中濃度が上昇する可能性がある。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 間質性肺疾患
11.1.2 体液貯留
浮腫(79.8%)、低アルブミン血症(38.1%)、胸水(9.5%)、心嚢液貯留(2.4%)等の体液貯留があらわれることがある。急激な体重の増加、呼吸困難等の異常が認められた場合には本剤の投与を中止するなど適切な処置を行うこと。
11.1.3 肝機能障害
血中ビリルビン増加(27.4%)、ALT増加(26.2%)、AST増加(21.4%)、ALP増加(8.3%)、γ-GTP増加(8.3%)、肝機能異常(4.8%)、肝損傷(1.2%)等の肝機能障害があらわれることがある。[8.2参照]
11.2 その他の副作用
| 10%以上 | 5%〜10%未満 | |
| 一般・全身障害及び投与部位の状態 | 疲労 | 倦怠感 |
| 代謝及び栄養障害 | 食欲減退(32.1%)、高血糖、低カリウム血症、高尿酸血症、低ナトリウム血症、低カルシウム血症 | |
| 臨床検査 | 洞性頻脈、体重減少、不整脈、血中尿素増加 | |
| 胃腸障害 | 悪心(28.6%)、嘔吐(23.8%)、便秘 | 上腹部痛、腹部膨満 |
| 血液及びリンパ系障害 | 貧血、白血球減少症、好中球減少症、血小板減少症 | |
| 腎及び尿路障害 | 血中クレアチニン増加、蛋白尿 | |
| 皮膚及び皮下組織障害 | 発疹 | |
| 筋骨格系及び結合組織障害 | 四肢痛 | 背部痛、筋肉痛 |
| 精神障害 | 不眠症 | |
| 呼吸器、胸郭及び縦隔障害 | 呼吸困難 | |
| 神経系障害 | 頭痛(32.1%) | 浮動性めまい |
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
16. 薬物動態
16.1 血中濃度
日本人の進行固形癌患者にグマロンチニブ200mg注)又は300mgを空腹時に単回経口投与及び1日1回反復経口投与したときのグマロンチニブのPKパラメータ及び血漿中濃度推移は以下のとおりであった1)。グマロンチニブ300mgを空腹時に1日1回反復経口投与したときの投与15日目におけるグマロンチニブの蓄積率はAUC0-24hが1.89、Cmaxが1.69であった。
注)本剤の承認用法・用量は「グマロンチニブとして1回300mgを1日1回空腹時に経口投与」である。
| 用量(mg) | 測定日(日) | 例数 | Cmax(ng/mL) | Tmax※(h) | AUC0-24h(ng・h/mL) |
| 200 | 1 | 3 | 2146(28) | 2.97(1.9-3.0) | 26936(26) |
| 15 | 3 | 3905(46) | 2.08(2.0-3.0) | 59726(34) | |
| 300 | 1 | 4 | 2199(23) | 1.95(1.0-3.0) | 30969(20) |
| 15 | 3 | 3838(55) | 1.93(1.0-2.0) | 61874(75) |
日本人患者にグマロンチニブ200mg又は300mgを単回経口投与(上図)及び1日1回反復経口投与(下図)したときの血漿中濃度推移(平均値±標準偏差)
単回経口投与
反復経口投与
16.2 吸収
16.2.1 食事の影響
16.3 分布
16.4 代謝
16.5 排泄
健康成人(6例)に14Cで標識されたグマロンチニブ300mgを単回経口投与したとき、投与240時間後までの糞中及び尿中に、それぞれ投与放射能の78.03%(未変化体として74.16%)及び20.34%(未変化体として0.02%)が排泄された6)(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 肝機能障害患者
グマロンチニブ300mgを単回経口投与したとき、肝機能正常被験者(76例)に対する軽度(20例)及び中等度(3例)の肝機能障害患者注)におけるグマロンチニブのCmax及びAUC0-24hの幾何平均値の比は、軽度の患者でそれぞれ1.04及び1.01、中等度の患者でそれぞれ0.896及びNC(算出せず)であった。[9.3参照]
注)NCI-ODWG(National Cancer Institute-Organ Dysfunction Working Group)基準による分類
16.7 薬物相互作用
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国際共同第II相試験(SCC244-108/GLORY試験)
MET遺伝子エクソン14スキッピング変異陽性の切除不能な進行・再発の非小細胞肺癌患者84例(日本人患者10例を含む)を対象に、本剤300mgを1日1回経口投与した8)9)。主要評価項目である独立画像判定機関の評価による奏効率(RECIST Ver1.1基準に基づく)は、有効性評価対象注)79例(日本人患者8例を含む)で65.8%(95%信頼区間:54.3-76.1)であった。
安全性評価対象84例中、副作用は97.6%(82/84例)に認められた。主な副作用は、浮腫79.8%(67/84例)、低アルブミン血症38.1%(32/84例)、頭痛32.1%(27/84例)、食欲減退32.1%(27/84例)、悪心28.6%(24/84例)、血中ビリルビン増加27.4%(23/84例)、ALT増加26.2%(22/84例)、嘔吐23.8%(20/84例)、AST増加21.4%(18/84例)であった。
安全性評価対象84例中、副作用は97.6%(82/84例)に認められた。主な副作用は、浮腫79.8%(67/84例)、低アルブミン血症38.1%(32/84例)、頭痛32.1%(27/84例)、食欲減退32.1%(27/84例)、悪心28.6%(24/84例)、血中ビリルビン増加27.4%(23/84例)、ALT増加26.2%(22/84例)、嘔吐23.8%(20/84例)、AST増加21.4%(18/84例)であった。
注)安全性評価対象のうち中央検査機関によりMET遺伝子エクソン14スキッピング変異陽性が確認された切除不能な進行・再発の非小細胞肺癌患者が有効性評価対象とされた。
18. 薬効薬理
18.1 作用機序
18.2 抗腫瘍作用
グマロンチニブは、MET遺伝子エクソン14スキッピング変異等を有する非小細胞肺癌患者由来腫瘍組織片を皮下移植したヌードマウスにおいて、腫瘍増殖抑制作用を示した12)。
19. 有効成分に関する理化学的知見
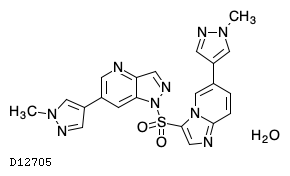
19.1. グマロンチニブ水和物
| 一般的名称 | グマロンチニブ水和物 |
|---|---|
| 一般的名称(欧名) | Gumarontinib Hydrate |
| 化学名 | 6-(1-Methyl-1H-pyrazol-4-yl)-1-[6-(1-methyl-1H-pyrazol-4-yl)imidazo[1,2-a]pyridine-3-sulfonyl]-1H-pyrazolo[4,3-b]pyridine monohydrate |
| 分子式 | C21H17N9O2S・H2O |
| 分子量 | 477.50 |
| 物理化学的性状 | 本品はほぼ白色〜白色の粉末である。 |
| KEGG DRUG |
|
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
PTP包装
24錠(8錠×3)
23. 主要文献
- 社内資料:SCC244-102試験(2024年6月24日承認、CTD2.7.2.2.3)
- 社内資料:SCC244-106試験(2024年6月24日承認、CTD2.7.2.2.4)
- 社内資料:薬物動態試験(2024年6月24日承認、CTD2.6.4.4.1)
- 社内資料:薬物動態試験(2024年6月24日承認、CTD2.6.4.4.2)
- 社内資料:薬物動態試験(2024年6月24日承認、CTD2.6.4.9)
- 社内資料:SCC244-107試験(2024年6月24日承認、CTD2.7.2.2.5)
- 社内資料:薬物相互作用(2024年6月24日承認、CTD2.6.4.7.4)
- 社内資料:SCC244-108国際共同第II相試験(2024年6月24日承認、CTD2.7.6.5.2)
- Yu Y,et al., eClinicalMedicine., 59, 101952, (2023) »PubMed
- 社内資料:薬効薬理試験(2024年6月24日承認、CTD2.6.2.2)
- Ai J,et al., Mol Cancer Ther., 17 (4), 751-762, (2018) »PubMed
- 社内資料:薬効薬理試験(2024年6月24日承認、CTD2.6.2.2.2.6)
24. 文献請求先及び問い合わせ先
文献請求先
大鵬薬品工業株式会社
医薬品情報課
〒101-8444
東京都千代田区神田錦町1-27
電話:0120-20-4527
製品情報問い合わせ先
大鵬薬品工業株式会社
医薬品情報課
〒101-8444
東京都千代田区神田錦町1-27
電話:0120-20-4527
26. 製造販売業者等
26.1 製造販売元
海和製薬株式会社
東京都港区赤坂5-2-33
26.2 販売元
大鵬薬品工業株式会社
東京都千代田区神田錦町1-27