医薬品情報
| 総称名 | ビヨントラ |
|---|---|
| 一般名 | アコラミジス塩酸塩 |
| 製剤名 | アコラミジス塩酸塩 |
| 薬効分類名 | トランスサイレチン型心アミロイドーシス治療薬 |
| 薬効分類番号 | 2190 |
| ATCコード | C01EB25 |
| KEGG DRUG |
D11973
アコラミジス塩酸塩
|
| JAPIC | 添付文書(PDF) |
添付文書情報2025年5月 改訂(第3版)

|
2.禁忌 4.効能または効果 5.効能又は効果に関連する注意 6.用法及び用量 8.重要な基本的注意 9.特定の背景を有する患者に関する注意 11.副作用 14.適用上の注意 16.薬物動態 17.臨床成績 18.薬効薬理 19.有効成分に関する理化学的知見 21.承認条件 22.包装 23.主要文献 24.文献請求先及び問い合わせ先 25.保険給付上の注意 26.製造販売業者等 |
商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| ビヨントラ錠400mg | BEYONTTRA tablets | アレクシオンファーマ | 2190048F1029 | 8979円/錠 | 処方箋医薬品注) |
2. 禁忌
次の患者には投与しないこと
本剤の成分に対し過敏症の既往歴のある患者
4. 効能または効果
トランスサイレチン型心アミロイドーシス(野生型及び変異型)
5. 効能または効果に関連する注意
5.1 本剤の適用にあたっては、最新のガイドライン等を参照し、トランスサイレチンアミロイドーシスの診断が確定していることを確認すること。
5.2 本剤は、トランスサイレチン型心アミロイドーシスによる心不全を有する患者に使用すること。また、「臨床成績」の項の内容を熟知し、臨床試験の選択基準等を十分理解した上で、適応患者の選択を行うこと。[17.1.1、17.1.2参照]
5.3 NYHA心機能分類III度の患者では、NYHA心機能分類I・II度の患者より相対的に本剤の有効性が低い可能性があるので、本剤の作用機序、及び臨床試験で示唆されたNYHA心機能分類と有効性の関係を十分に理解し、患者の状態を考慮した上で、本剤投与の要否を判断すること。[17.1.1参照]
5.4 NYHA心機能分類IV度の患者における有効性及び安全性は確立していない。
5.5 肝移植後の患者における有効性及び安全性は確立していない。
6. 用法及び用量
通常、成人にはアコラミジス塩酸塩として1回800mgを1日2回経口投与する。
8. 重要な基本的注意
8.1 本剤の投与開始初期に、eGFRが低下することがあることから、腎機能を定期的に検査すること。腎機能障害のある患者では経過を十分に観察し、腎機能障害の悪化に注意すること。[9.2.1参照]
9. 特定の背景を有する患者に関する注意
9.2 腎機能障害患者
9.2.1 重度の腎機能障害患者又は末期腎不全患者
投与の必要性を慎重に判断すること。本剤投与によりeGFRが低下することがあり、腎機能が悪化するおそれがある。eGFRが15mL/min/1.73m2未満の患者は、臨床試験では除外されている。[8.1参照]
9.3 肝機能障害患者
9.3.1 中等度又は重度の肝機能障害患者
本剤は主に胆汁中に排泄されるため、血中濃度が上昇するおそれがある。AST、ALT又は総ビリルビンが基準値上限の3倍を超える患者は、臨床試験では除外されている。
9.5 妊婦
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。動物実験(ラット)の結果から、本剤は乳汁中に移行する可能性がある。[9.5参照]
9.7 小児等
小児等を対象とした臨床試験は実施していない。
11. 副作用
11.2 その他の副作用
| 1%以上2%未満 | 0.5%以上1%未満 | |
| 胃腸障害 | 悪心 | 下痢、腹部不快感、上腹部痛 |
| 臨床検査 | − | 血中クレアチニン増加 |
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
16. 薬物動態
16.1 血中濃度
16.1.1 単回投与
日本人健康成人9例に本剤400mg注)及び800mgを絶食下で単回経口投与したときのアコラミジスの血漿中濃度推移及び薬物動態パラメータは以下のとおりであった2)。
| 投与量 | Cmax(ng/mL) | Tmaxa)(h) | AUC0-inf(ng・h/mL) | T1/2(h) |
| 400mg | 6621(2342.7) | 1.0[0.5,1.0] | 106992(33400.1) | 34(12.5) |
| 800mg | 11699(5766.9) | 1.0[0.5,2.0] | 177305(83313.2) | 44(27.8) |
注)本剤の承認された用法及び用量は、1回800mgを1日2回経口投与である。
16.1.2 反復投与
健康成人6例に本剤800mgを絶食下で1日2回12日間経口投与したときのアコラミジスの血漿中濃度は、投与10日目までに定常状態に達し、Cmaxに基づく累積係数は1.3〜1.6であった3)(外国人データ)。
日本人の野生型又は変異型のトランスサイレチン型心アミロイドーシス患者に本剤800mgを1日2回経口投与したときの定常状態における薬物動態パラメータの母集団薬物動態解析に基づく推定値は以下のとおりであった2)。
| 投与量 | Cmax,ss(ng/mL) | Ctrough,ss(ng/mL) | AUC0-12h,ss(ng・h/mL) |
| 800mg | 15768(1650) | 2638(1429) | 55486(18761) |
16.2 吸収
16.2.1 食事の影響
健康成人17例に本剤800mgを空腹時又は高脂肪食摂取後に単回経口投与したとき、食事によりアコラミジスのCmaxは約22%低下したが、AUC0-infは食事の影響を受けなかった4)(外国人データ)。
16.3 分布
アコラミジスのヒト血漿タンパク結合率は96.3%〜96.5%であった3)(in vitroデータ)。
16.4 代謝
健康成人6例に本剤の14C標識体800mgを単回経口投与したとき、アコラミジスは主にグルクロン酸抱合による代謝で消失した。ヒト血漿中総放射能の約63.1%が未変化体、約7.6%がグルクロン酸抱合体であった3)(外国人データ)。
16.5 排泄
健康成人6例に本剤の14C標識体800mgを単回経口投与したとき、投与後216時間までの糞中及び尿中に投与量のそれぞれ約34%及び約68%が排泄された。未変化体の尿中排泄率は投与量の10%以下であった6)(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害
健康成人及びATTR-CM患者330例(日本人33例)を対象とした母集団薬物動態解析の結果、腎機能(クレアチニンクリアランス:25.9〜190mL/min、eGFR:25.4〜157mL/min/1.73m2)はアコラミジスの見かけのクリアランスに影響を及ぼさなかった3)。
16.6.2 高齢者
健康成人及びATTR-CM患者330例(日本人33例)を対象とした母集団薬物動態解析の結果、年齢(18〜89歳)はアコラミジスの見かけのクリアランス及び分布容積に影響を及ぼさなかった3)。
16.7 薬物相互作用
16.7.1 In vitro試験
16.7.2 その他の薬剤
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 海外第III相試験(AG10-301)
野生型又は変異型のトランスサイレチン型心アミロイドーシス患者注)632例(プラセボ群211例、本剤群421例)を対象とした30ヵ月間の二重盲検プラセボ対照試験*1を実施した。本剤の用法・用量は800mg 1日2回とした。このうち、ベースライン時のeGFRが30mL/min/1.73m2以上の患者611例(プラセボ群202例、本剤群409例)が有効性の主要な解析対象集団とされた。
投与開始12ヵ月後以降はタファミジスメグルミン又はタファミジスを併用可能とされ、併用割合はプラセボ群22.8%(46例)、本剤群14.9%(61例)であった。
Part Bの主要評価項目である「投与開始30ヵ月後までの死因を問わない死亡割合、心血管事象に関連する入院頻度、脳性ナトリウム利尿ペプチド前駆体N端フラグメント(NT-proBNP)のベースラインからの変化量、及び6分間歩行距離のベースラインからの変化量から構成される階層的複合エンドポイント」について、本剤群でプラセボ群と比べて統計学的に有意な差が認められた(p<0.0001(有意水準4%(両側)*2)、Finkelstein-Schoenfeld法)。主要評価項目の構成要素別の結果を下表に示す。
| プラセボ群 (202例) | 本剤群 (409例) | |
| 死因を問わない死亡 (発現割合%(発現例数)) | 25.7(52) | 19.3(79) |
| 生存例における心血管事象に関連する入院頻度(回/年) 平均値±標準偏差(発現例数/対象例数) | 0.293±0.5751(50/150) | 0.132±0.3257(68/330) |
| NT-proBNP(pg/mL) | ||
| ベースライン値 | 2650.11±1899.482(202) | 2865.33±2149.639(409) |
| 投与開始30ヵ月後の測定値 | 4348.33±4758.130(133) | 2853.70±2876.953(280) |
| ベースラインに対する比 調整幾何平均値[95%信頼区間]a | 2.771[2.485,3.091] | 1.465[1.356,1.583] |
| 6分間歩行距離(m) | ||
| ベースライン値 | 351.51±93.828(202) | 362.78±103.501(407) |
| 投与開始30ヵ月後の測定値 | 322.38±120.916(121) | 365.96±124.734(269) |
| ベースラインからの変化量 最小二乗平均値[95%信頼区間]a | −104.29[−119.53,−89.06] | −64.65[−75.45,−53.86] |
また、投与開始30ヵ月後までの死因を問わない死亡割合及び心血管事象に関連する入院頻度のNYHA心機能分類(I/IIとIII)別の部分集団解析の結果、死因を問わない死亡割合(発現例数)はNYHA心機能分類I/II度の集団でプラセボ群24.3%(42/173例)、本剤群17.4%(59/339例)(ハザード比[95%信頼区間]*3:0.695[0.466,1.036])、NYHA心機能分類III度の集団でプラセボ群34.5%(10/29例)、本剤群28.6%(20/70例)(ハザード比[95%信頼区間]*3:1.145[0.515,2.546])、生存例における心血管事象に関連する入院頻度(発現例数/対象例数)は、NYHA心機能分類I/II度の集団でプラセボ群0.31回/年(47/131例)、本剤群0.12回/年(55/280例)、NYHA心機能分類III度の集団でプラセボ群0.18回/年(3/19例)、本剤群0.21回/年(13/50例)、全症例における心血管事象に関連する入院頻度(発現例数/対象例数)は、NYHA心機能分類I/II度の集団でプラセボ群0.51回/年(74/173例)、本剤群0.25回/年(87/339例)、NYHA心機能分類III度の集団でプラセボ群0.75回/年(12/29例)、本剤群0.47回/年(22/70例)であった7)8)。
副作用発現頻度は本剤群で11.9%(50/421例)、プラセボ群で5.2%(11/211例)であった。本剤群の主な副作用として、悪心[本剤群1.4%(6/421例)、プラセボ群0.5%(1/211例)、以下同順]、下痢[1.0%(4/421例)、0%]、発疹[1.0%(4/421例)、0.5%(1/211例)]が認められた9)。[5.2、5.3参照]
副作用発現頻度は本剤群で11.9%(50/421例)、プラセボ群で5.2%(11/211例)であった。本剤群の主な副作用として、悪心[本剤群1.4%(6/421例)、プラセボ群0.5%(1/211例)、以下同順]、下痢[1.0%(4/421例)、0%]、発疹[1.0%(4/421例)、0.5%(1/211例)]が認められた9)。[5.2、5.3参照]
*1:二重盲検投与期間のうち投与開始から12ヵ月間がPart A、二重盲検投与期間全体がPart Bとされ、早期承認申請を目的とし検討したPart Aの主要評価項目(6分間歩行距離のベースラインから投与開始12ヵ月後までの変化量)について、プラセボ群と本剤群で統計学的に有意な差は認められなかった。
*2:試験全体の第一種の過誤確率を制御するためにPart Bの主要評価項目の解析に用いる有意水準を4%(両側)とした。
*3:投与群、ベースライン時の6分間歩行距離、ベースライン時のNYHA心機能分類(I/II度/III度)、ベースラインのNYHA心機能分類とベースライン時の6分間歩行距離の交互作用を因子として、病型(野生型/変異型)、スクリーニング時のNT-proBNP(3000pg/mL以下/超)、スクリーニング時のeGFR(45mL/min/1.73m2未満/以上)を層とした層別Cox比例ハザードモデル
17.1.2 国内第III相試験(ALXN2060-TAC-302)
注)主な選択基準は以下のとおりであった。
・以下のいずれかの検査及び遺伝子検査により、野生型又は変異型のトランスサイレチン型心アミロイドーシスと確定診断された患者
a)心内膜心筋生検で、アミロイドタイピング(免疫組織染色法、質量分析法又は免疫電子顕微鏡法)によりTTRアミロイド沈着が確認されている
b)99mTc-ピロリン酸又は99mTc-ビスホスホネートシンチグラフィ(DPD又はHMDP)により陽性像が確認され、かつ血清及び尿免疫固定法、並びに血清免疫グロブリン遊離軽鎖測定に基づきALアミロイドーシスが除外されている*4
(*4:ALXN2060-TAC-302試験のみ、心臓以外の生検(腹壁脂肪等)によるTTRアミロイド沈着の確認も必要)
・NYHA心機能分類I〜III度で、以下のいずれかの心不全の病歴を有する患者
a)心不全による入院歴を有する
b)心不全による入院歴はないが、容量負荷や心内圧上昇の徴候・症状による心不全の臨床的エビデンス(頚静脈圧上昇、息切れ、X線検査又は聴診での肺うっ血の徴候、末梢浮腫等)を有する
c)利尿薬による治療を必要とした又は継続的に必要とする心不全の症状を有する
・スクリーニング前10年以内に受けた経胸壁心エコー図検査又は心臓MRIによる左室壁(心室中隔又は左室後壁)の厚さが12mm以上である患者
18. 薬効薬理
18.1 作用機序
アコラミジスはTTRの4量体における2つのサイロキシン結合部位に選択的に結合することにより、疾患抑制性のT119M変異型TTRと同様の立体構造にシフトすることで4量体を安定化させ、その単量体への解離を抑制し、新たなTTRアミロイド形成の律速段階を抑制する。
18.1.1 精製TTRへの結合
(1)蛍光偏光法、等温滴定熱量測定法、表面プラズモン共鳴法、マイクロスケール熱泳動法、サブユニット交換法によるin vitro試験において、本剤はTTRに対して結合親和性を示した10)。
(2)表面プラズモン共鳴法によるin vitro試験において、本剤はTTRに速やかに結合と緩やかな遊離を示した10)。
(3)線維形成法によるin vitro試験において、本剤は野生型TTR及びV122I変異型TTRを安定化した10)。
18.1.2 血清TTRへの結合
(1)12種の固有のTTR変異を含む54例の被験者血清を用いた蛍光プローブ排除法による評価で、本剤は検討されたすべての変異型TTR及び対照に用いた野生型TTRに対し、90%超のTTR結合率を示した10)。
(2)18種の固有のTTR変異を含む64例の被験者血漿TTRを用いたウエスタンブロット法による評価で、本剤は検討されたほとんどの変異型TTR及び対照に用いた野生型TTRに対し、90%超のTTR安定化率を示した10)。
18.1.3 TTR結合時の立体構造
結晶構造による解析で、本剤は野生型TTR及びV122I変異型TTRに結合すると、Ser117側鎖への相互作用により、疾患抑制性のT119M変異型TTRと同様の立体構造にシフトすることが示された10)。
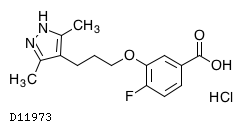
19. 有効成分に関する理化学的知見
19.1. アコラミジス塩酸塩
| 一般的名称 | アコラミジス塩酸塩 |
|---|---|
| 化学名 | 3-[3-(3,5-Dimethyl-1H-pyrazol-4-yl)propoxy]-4-fluorobenzoic acid monohydrochloride |
| 分子式 | C15H17FN2O3・HCl |
| 分子量 | 328.77 |
| 物理化学的性状 | 白色から褐色の固体である。エタノールにやや溶けにくく、水に溶けにくい。 |
| 分配係数 | Log D=0.52(pH 7) |
| KEGG DRUG |
|
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
56錠[4錠(PTP)×14]
23. 主要文献
- 社内資料:毒性試験の概要文(2025年3月27日承認、CTD2.6.6.9)
- 社内資料:アコラミジスの薬物動態に関する資料
- 社内資料:臨床薬理の概要(2025年3月27日承認、CTD2.7.2.2、CTD2.7.2.3)
- 社内資料:生物薬剤学試験及び関連する分析法の概要(2025年3月27日承認、CTD2.7.1.3)
- 社内資料:薬物動態試験の概要文(2025年3月27日承認、CTD2.6.4.7)
- 社内資料:健康成人男性を対象に[14C]-アコラミジスを経口投与したときの吸収、代謝、排泄及びマスバランスを評価する海外第I相試験(AG10-007試験)(2025年3月27日承認、CTD2.7.6.4)
- 社内資料:臨床的有効性の概要(2025年3月27日承認、CTD2.7.3.2.1、CTD2.7.3.2.2)
- Gillmore JD,et al., N Engl J Med, 390, 132-142, (2024) »PubMed
- 社内資料:臨床的安全性の概要(2025年3月27日承認、CTD2.7.4.2.1)
- 社内資料:効力を裏付ける試験(2025年3月27日承認、CTD2.6.2.2)
24. 文献請求先及び問い合わせ先
文献請求先
アレクシオンファーマ合同会社
メディカル インフォメーション センター
〒108-0023
東京都港区芝浦三丁目1番1号 田町ステーションタワーN
電話:0120-577-657
製品情報問い合わせ先
アレクシオンファーマ合同会社
メディカル インフォメーション センター
〒108-0023
東京都港区芝浦三丁目1番1号 田町ステーションタワーN
電話:0120-577-657
25. 保険給付上の注意
本剤は新医薬品であるため、厚生労働省告示第107号(平成18年3月6日付)に基づき、2026年5月末日までは、投薬は1回14日分を限度とされている。
26. 製造販売業者等
26.1 製造販売元
アレクシオンファーマ合同会社
〒108-0023
東京都港区芝浦三丁目1番1号 田町ステーションタワーN