医薬品情報
| 総称名 | インレビック |
|---|---|
| 一般名 | フェドラチニブ塩酸塩水和物 |
| 欧文一般名 | Fedratinib Hydrochloride Hydrate |
| 製剤名 | フェドラチニブ塩酸塩水和物カプセル |
| 薬効分類名 | 抗悪性腫瘍剤 ヤヌスキナーゼ(JAK)阻害剤 |
| 薬効分類番号 | 4291 |
| ATCコード | L01EJ02 |
| KEGG DRUG |
D11296
フェドラチニブ塩酸塩水和物
|
| KEGG DGROUP |
DG02020
JAK阻害薬
|
| JAPIC | 添付文書(PDF) |
添付文書情報2026年5月 改訂(第3版)

商品情報 3.組成・性状
| 販売名 | 欧文商標名 | 製造会社 | YJコード | 薬価 | 規制区分 |
|---|---|---|---|---|---|
| インレビックカプセル100mg | Inrebic capsules | レコルダティ・レア・ディジーズ・ジャパン | 4291099M1022 | 11137.4円/カプセル | 劇薬, 処方箋医薬品 |
1. 警告
1.1 本剤は、緊急時に十分対応できる医療施設において、造血器悪性腫瘍の治療に対して十分な知識・経験を持つ医師のもとで、本剤の投与が適切と判断される症例についてのみ投与すること。また、治療開始に先立ち、患者又はその家族に有効性及び危険性を十分に説明し、同意を得てから投与を開始すること。
2. 禁忌
次の患者には投与しないこと
本剤の成分に対し過敏症の既往歴のある患者
4. 効能または効果
5. 効能または効果に関連する注意
5.1 臨床試験に組み入れられた患者のリスク分類、脾臓の大きさ等について、「17.臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分理解した上で、適応患者の選択を行うこと。[17.1.1-17.1.3参照]
5.2 病理組織学的検査を行い、骨髄線維症と診断された患者に使用すること。
6. 用法及び用量
通常、成人にはフェドラチニブとして1回400mgを1日1回経口投与する。なお、患者の状態により適宜減量する。
7. 用法及び用量に関連する注意
7.1 他の抗悪性腫瘍剤との併用について、有効性及び安全性は確立していない。
7.2 本剤投与により副作用が発現した場合には、以下の基準を参考に休薬、減量又は中止すること。また、200mgで忍容性が得られない場合は、本剤の投与を中止すること。
| 段階 | 本剤投与量 |
| 用量レベル1 | 400mg |
| 用量レベル2 | 300mg |
| 用量レベル3 | 200mg |
| 副作用注1) | 処置 |
| 出血を伴うGrade3の血小板数減少、Grade4の血小板数減少 | Grade2以下又はベースラインに回復するまで休薬する。回復後、休薬前の投与量から1用量レベル下げて投与再開する。 |
| Grade4の好中球数減少 | Grade2以下又はベースラインに回復するまで休薬する。回復後、休薬前の投与量から1用量レベル下げて投与再開する。 |
| Grade3以上の赤血球輸血を要する貧血 | Grade2以下又はベースラインに回復するまで休薬する。回復後、休薬前の投与量から1用量レベル下げて投与再開する。 |
| Grade3以上の悪心、嘔吐、下痢で、対症療法を行っても48時間以内に回復しない場合 | Grade1以下又はベースラインに回復するまで休薬する。回復後、休薬前の投与量から1用量レベル下げて投与再開する。 |
| Grade3以上のALT、AST、ビリルビン増加 | ・Grade1以下又はベースラインに回復するまで休薬する。回復後、休薬前の投与量から1用量レベル下げて投与再開する。 ・Grade3以上が再発した場合には、投与を中止する。 |
| 脳症を疑う神経学的所見 | ・Grade1以下又はベースラインに回復するまで休薬する注2)。回復後、休薬前の投与量から1用量レベル下げて投与再開する。 ・ウェルニッケ脳症の場合には、投与を中止する。 |
| Grade3以上の上記以外の非血液系の副作用 | Grade1以下又はベースラインに回復するまで休薬する。回復後、休薬前の投与量から1用量レベル下げて投与再開する。 |
8. 重要な基本的注意
8.1 ビタミンB1(チアミン)の欠乏によりウェルニッケ脳症があらわれることがあるので、本剤の投与にあたっては、以下の事項に注意すること。[1.2、7.3、11.1.1参照]
8.1.1 本剤の投与開始前にビタミンB1濃度を測定すること。ビタミンB1の減少が認められる患者に対してはビタミンB1補充を行い、ビタミンB1濃度が回復するまで本剤投与を開始しないこと。
8.1.2 本剤投与中はビタミンB1経口剤の投与を行い、ビタミンB1欠乏症の症状又は徴候が認められる場合など、必要に応じてビタミンB1濃度の測定を行うこと。
8.1.3 嘔吐、下痢等からビタミンB1欠乏を含む低栄養状態等の悪化を引き起こす可能性があるため、制吐剤又は止瀉剤の予防投与を検討すること。
8.1.4 神経内科医との連携の下、神経学的症状を含む患者の状態を注意深く観察すること。
8.2 免疫抑制作用により、細菌、真菌、ウイルス又は原虫による感染症や日和見感染が発現又は悪化することがある。肝炎ウイルス、結核等が再活性化するおそれがあるので、本剤投与に先立って肝炎ウイルス、結核等の感染の有無を確認し、本剤の投与開始前に適切な処置の実施を考慮すること。本剤投与中は感染症の発現又は増悪に十分注意すること。[1.3、9.1.1-9.1.3、11.1.2参照]
8.3 骨髄抑制があらわれることがあるので、本剤の投与開始前及び投与中は定期的に血液検査(血球数算定、白血球分画等)を行い、患者の状態を十分に観察すること。[11.1.3参照]
8.4 肝機能障害があらわれることがあるので、本剤の投与開始前及び投与中は定期的に肝機能検査を行うこと。[11.1.5参照]
8.5 ぶどう膜炎があらわれることがあるので、眼の異常の有無を定期的に確認すること。また、眼の異常が認められた場合には、速やかに医療機関を受診するよう患者を指導すること。[11.1.7参照]
9. 特定の背景を有する患者に関する注意
9.1 合併症・既往歴等のある患者
9.1.1 結核の既感染者(特に結核の既往歴のある患者及び胸部レントゲン上結核治癒所見のある患者)
9.1.2 感染症(敗血症、肺炎、ウイルス感染等)を合併している患者
9.1.3 B型肝炎ウイルスキャリアの患者又は既往感染者(HBs抗原陰性、かつHBc抗体又はHBs抗体陽性)
9.2 腎機能障害患者
9.2.1 重度の腎機能障害のある患者(CLcr 15mL/min以上30mL/min未満)
9.2.2 中等度の腎機能障害のある患者(CLcr 30mL/min以上60mL/min未満)
患者の状態をより慎重に観察し、副作用の発現に十分注意すること。本剤の血中濃度が上昇し、副作用が強くあらわれるおそれがある。[16.6.1参照]
9.3 肝機能障害患者
9.3.1 重度の肝機能障害のある患者(Child-Pugh分類C)
患者の状態をより慎重に観察し、副作用の発現に十分注意すること。本剤の血中濃度が上昇し、副作用が強くあらわれるおそれがある。[16.6.2参照]
9.4 生殖能を有する者
妊娠する可能性のある女性には、本剤投与中及び最終投与後1ヵ月間において避妊する必要性及び適切な避妊法について説明すること。[9.5参照]
9.5 妊婦
9.6 授乳婦
授乳しないことが望ましい。本剤が乳汁に移行する可能性があり、乳児が乳汁を介して本剤を摂取した場合、乳児に重篤な副作用が発現するおそれがある。
9.7 小児等
小児等を対象とした臨床試験は実施していない。
10. 相互作用
相互作用序文
本剤は、主にCYP3Aで代謝され、一部はCYP2C19によっても代謝される。また、本剤はCYP3A、CYP2C19、CYP2D6、OCT2、MATE1及びMATE2-Kに対して阻害作用を示す。
薬物代謝酵素用語
CYP3A
薬物代謝酵素用語
CYP2C19
薬物代謝酵素用語
CYP2D6
薬物代謝酵素用語
OCT2
薬物代謝酵素用語
MATE1
薬物代謝酵素用語
MATE2-K
10.2 併用注意
| 強いCYP3A阻害剤 リトナビル、イトラコナゾール、クラリスロマイシン等 [7.5、16.7.1参照] | 本剤の副作用が増強されるおそれがあるので、これらの薬剤との併用は可能な限り避け、強いCYP3A阻害作用のない薬剤への代替を考慮すること。やむを得ず併用する場合には、本剤を減量するとともに、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | これらの薬剤がCYP3Aを阻害することにより、本剤の血中濃度が上昇する可能性がある。 |
| 中程度のCYP3Aかつ強いCYP2C19阻害剤 フルコナゾール [16.7.2参照] | 本剤の副作用が増強されるおそれがあるので、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | フルコナゾールがCYP3A及びCYP2C19を同時に阻害することにより、本剤の血中濃度が上昇する可能性がある。 |
| 強い又は中程度のCYP3A誘導剤 リファンピシン、フェニトイン、カルバマゼピン等 [16.7.3、16.7.4参照] | 本剤の有効性が減弱するおそれがあるので、CYP3A誘導作用のない薬剤への代替を考慮すること。 | これらの薬剤がCYP3Aを誘導することにより、本剤の血中濃度が低下する可能性がある。 |
| CYP3Aの基質となる薬剤 ミダゾラム、フェンタニル、トリアゾラム等 [16.7.5参照] | これらの薬剤の副作用が増強されるおそれがあるので、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | 本剤がCYP3Aを阻害することにより、これらの薬剤の血中濃度が上昇する可能性がある。 |
| CYP2C19の基質となる薬剤 オメプラゾール、ランソプラゾール、ジアゼパム等 [16.7.5参照] | これらの薬剤の副作用が増強されるおそれがあるので、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | 本剤がCYP2C19を阻害することにより、これらの薬剤の血中濃度が上昇する可能性がある。 |
| CYP2D6の基質となる薬剤 メトプロロール、アミトリプチリン、ペルフェナジン等 [16.7.5参照] | これらの薬剤の副作用が増強されるおそれがあるので、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | 本剤がCYP2D6を阻害することにより、これらの薬剤の血中濃度が上昇する可能性がある。 |
| OCT2、MATE1及びMATE2-Kの基質となる薬剤 メトホルミン、プロカインアミド等 [16.7.6参照] | これらの薬剤の副作用が増強されるおそれがあるので、患者の状態を慎重に観察し、副作用の発現に十分注意すること。 | 本剤がOCT2、MATE1及びMATE2-Kを阻害することにより、これらの薬剤の腎クリアランスが低下する可能性がある。 |
11. 副作用
11.1 重大な副作用
次の副作用があらわれることがあるので、観察を十分に行い、異常が認められた場合には投与を中止するなど適切な処置を行うこと。
11.1.1 脳症(ウェルニッケ脳症含む)(0.5%)
11.1.2 感染症
11.1.3 骨髄抑制
貧血(36.2%)、血小板減少(20.8%)、好中球減少(5.4%)があらわれることがある。[8.3参照]
11.1.4 出血
血栓性血小板減少性紫斑病(0.5%)、上部消化管出血(0.5%)等があらわれることがある。
11.1.5 肝機能障害
AST(3.6%)、ALT(7.2%)増加を伴う肝機能障害があらわれることがある。[8.4参照]
11.1.6 間質性肺疾患(頻度不明)
11.1.7 ぶどう膜炎(0.9%)[8.5参照]
11.2 その他の副作用
| 5%以上 | 5%未満 | 頻度不明 | |
| 胃腸 | 下痢(52.0%)、悪心(50.2%)、嘔吐(35.7%)、腹痛、便秘 | 膵炎、消化不良、上腹部痛 | |
| 神経系 | 頭痛 | 浮動性めまい、味覚不全 | |
| 皮膚 | そう痒症 | ||
| 代謝 | リパーゼ増加 | アミラーゼ増加、高カリウム血症 | |
| 腎 | 血中クレアチニン増加 | 排尿困難 | |
| 筋・骨格系 | 筋痙縮、骨痛、四肢痛 | ||
| 呼吸器 | 呼吸困難 | ||
| 感染症 | 尿路感染 | ||
| 血管 | 高血圧 | ||
| その他 | 疲労 | 体重増加、無力症 |
14. 適用上の注意
14.1 薬剤交付時の注意
PTP包装の薬剤はPTPシートから取り出して服用するよう指導すること。PTPシートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
15. その他の注意
15.1 臨床使用に基づく情報
心血管系事象のリスク因子を有する関節リウマチ患者を対象としたJAK阻害剤トファシチニブクエン酸塩の海外臨床試験の結果、主要評価項目である主要な心血管系事象(Major Adverse Cardiovascular Events:MACE)及び悪性腫瘍(非黒色腫皮膚癌を除く)の発現率について、TNF阻害剤群に対するハザード比(95%信頼区間)はそれぞれ1.33(0.91,1.94)及び1.48(1.04,2.09)であり、95%信頼区間上限は予め設定していた非劣性マージン1.8を超え、TNF阻害剤群に対する非劣性が検証されなかったことが報告されている。
15.2 非臨床試験に基づく情報
イヌの28日間反復投与毒性試験において、臨床曝露量を下回る用量から精巣上体・精巣の無精子症・乏精子症、精細管変性が認められた。
16. 薬物動態
16.1 血中濃度
16.1.1 単回及び反復投与
日本人の骨髄線維症患者に本剤300注1)及び400mgを1日1回反復経口投与したときの、初回投与後及び反復投与28日目のフェドラチニブの薬物動態パラメータは下表のとおりであった3)。
| 用量(mg) | 測定日(日) | 例数 | Cmax(ng/mL) | Tmax(h) | AUCtau(ng・h/mL) |
| 300注1) | 1 | 3 | 1139(42) | 2.02(2.00,3.02) | 8015(59) |
| 28 | 3 | 1685(6) | 3.00(2.00,4.00) | 24710(20) | |
| 400 | 1 | 3 | 3324(21) | 3.00(1.95,6.08) | 22875(15) |
| 28 | 3 | 3698(49) | 2.67(0.95,4.00) | 53816(104) |
また、日本人を含む骨髄線維症患者から得られた血漿中濃度データを用いて、母集団薬物動態解析を実施した。本剤400mgを1日1回反復経口投与したときの、定常状態におけるフェドラチニブの日本人の薬物動態パラメータは下表のとおりであった4)。
| CL/F(L/h) | AUC0-24h(ng・h/mL) | Cmax(ng/mL) | Ctrough(ng/mL) | t1/2(h) | Tmax(h) |
| 9.6(51.3) | 41316(51.3) | 2417(53.5) | 1418(58.1) | 144.8(38.3) | 2(1,7) |
16.2 吸収
16.2.1 食事の影響
健康成人(19例)に本剤500mg注1)を単回経口投与したとき、空腹時投与に対する低脂肪・低カロリー食及び高脂肪・高カロリー食摂取後投与における、フェドラチニブのCmaxの幾何平均値の比はそれぞれ1.12及び0.91、AUCinfの幾何平均値の比はそれぞれ1.22及び1.19であった5)(外国人データ)。
16.3 分布
16.4 代謝
フェドラチニブは主としてCYP3Aで代謝され、またCYP3Aに比べて寄与率は小さいがCYP2C19及びフラビン含有モノオキシゲナーゼ3によっても代謝されると考えられる8)。
16.5 排泄
健康成人(6例)に14C標識したフェドラチニブ200mg注1)を単回経口投与したとき、放射能の総回収率は82.1%で、尿及び糞便中にそれぞれ5.15%及び76.9%が回収された。尿及び糞便中に回収された放射能に占める未変化体の割合はそれぞれ投与量の2.9%及び23.3%であった9)(外国人データ)。
16.6 特定の背景を有する患者
16.6.1 腎機能障害患者
16.6.2 肝機能障害患者
16.7 薬物相互作用
16.7.1 ケトコナゾール(強いCYP3A阻害剤)
16.7.2 フルコナゾール(中程度のCYP3Aかつ強いCYP2C19阻害剤)
16.7.3 リファンピシン(強いCYP3A誘導剤)
16.7.4 エファビレンツ(中程度のCYP3A誘導剤)
16.7.5 ミダゾラム(CYP3Aの基質)、オメプラゾール(CYP2C19の基質)、メトプロロール(CYP2D6の基質)
16.7.6 ジゴキシン(P-gpの基質)、ロスバスタチン(OATP1B1/1B3及びBCRPの基質)、メトホルミン(OCT2、MATE1及びMATE2-Kの基質)
16.7.7 生理学的薬物動態モデルによるシミュレーション(エリスロマイシン(中程度のCYP3A阻害剤))
生理学的薬物動態モデルによるシミュレーション結果から、健康成人にエリスロマイシン500mgを1日3回反復投与し、本剤400mgを併用で1日1回単回及び反復投与したとき、本剤単独投与時に対する併用投与時におけるフェドラチニブのAUCはそれぞれ1.85及び1.18と推定された13)。
16.8 その他
フェドラチニブはP-gpの基質である(in vitro)18)。
注1)本剤の承認された用法及び用量は、「通常、成人にはフェドラチニブとして1回400mgを1日1回経口投与する。なお、患者の状態により適宜減量する。」である。
注2)経口剤は国内未承認
17. 臨床成績
17.1 有効性及び安全性に関する試験
17.1.1 国内第I/II相試験(FEDR-MF-003試験)
JAK阻害剤による治療歴の有無を問わない骨髄線維症注1)患者を対象とした非盲検非対照試験を実施した19)。本剤400mgが1日1回経口投与注2)された第I相パートの3例及び第II相パートの25例(合計28例)において、主要評価項目である第6サイクル終了時点の脾臓容積がベースラインから35%以上縮小(SVR35)を達成した被験者の割合[95%信頼区間]は71.4%[51.3,86.8](20/28例)であった(2023年10月5日データカットオフ)。
副作用発現頻度は96.4%(27/28例)であった。主な副作用は、貧血60.7%(17/28例)、下痢及び血小板減少症各32.1%(9/28例)等であった。[5.1、5.3参照]
副作用発現頻度は96.4%(27/28例)であった。主な副作用は、貧血60.7%(17/28例)、下痢及び血小板減少症各32.1%(9/28例)等であった。[5.1、5.3参照]
17.1.2 海外第III相試験(EFC12153試験(JAKARTA試験))
JAK阻害剤による治療歴のない骨髄線維症注1)患者を対象とした二重盲検無作為化比較試験を実施した20)。本剤400mg群及びプラセボ群に、それぞれ96例が無作為に割付けられた(合計192例)。本剤400mgを1日1回経口投与注2)又はプラセボを1日1回経口投与した。
主要評価項目である第6サイクル終了時点及びその4週間後にSVR35を達成した被験者の割合[95%信頼区間]は本剤400mg群で36.5%[26.8,46.1](35/96例)、プラセボ群で1.0%[0,3.1](1/96例)であり、プラセボ群に対する本剤群の優越性が認められた(群間差[97.5%信頼区間]:35.4%[24.2,46.7]、カイ二乗検定p<0.0001、有意水準両側0.025(Bonferroni補正による調整))。
副作用発現頻度は、本剤400mg群で89.6%(86/96例)であった。主な副作用は、下痢58.3%(56/96例)、悪心58.3%(56/96例)、嘔吐40.6%(39/96例)、貧血34.4%(33/96例)等であった。[5.1、5.3参照]
主要評価項目である第6サイクル終了時点及びその4週間後にSVR35を達成した被験者の割合[95%信頼区間]は本剤400mg群で36.5%[26.8,46.1](35/96例)、プラセボ群で1.0%[0,3.1](1/96例)であり、プラセボ群に対する本剤群の優越性が認められた(群間差[97.5%信頼区間]:35.4%[24.2,46.7]、カイ二乗検定p<0.0001、有意水準両側0.025(Bonferroni補正による調整))。
副作用発現頻度は、本剤400mg群で89.6%(86/96例)であった。主な副作用は、下痢58.3%(56/96例)、悪心58.3%(56/96例)、嘔吐40.6%(39/96例)、貧血34.4%(33/96例)等であった。[5.1、5.3参照]
17.1.3 海外第II相試験(ARD12181試験(JAKARTA2試験))
ルキソリチニブによる治療歴のある骨髄線維症注1)患者を対象とした非盲検非対照試験を実施した21)。本剤400mgを開始用量として1日1回経口投与注2)し、投与開始後8週及び16週終了時に効果不十分と判定され、安全性が管理可能と判断された場合には、1日100mgずつ最大600mgまでの増量注3)が許容された。
評価可能であった83例において、主要評価項目である第6サイクル終了時点のSVR35を達成した被験者の割合(LOCF注4)法を用いた解析)[95%信頼区間]は48.2%[37.1,59.4](40/83例)であった。
副作用発現頻度は全投与例において90.7%(88/97例)であった。主な副作用は、悪心52.6%(51/97例)、下痢51.5%(50/97例)、嘔吐37.1%(36/97例)、貧血29.9%(29/97例)、血小板減少症20.6%(20/97例)等であった。[5.1、5.3参照]
評価可能であった83例において、主要評価項目である第6サイクル終了時点のSVR35を達成した被験者の割合(LOCF注4)法を用いた解析)[95%信頼区間]は48.2%[37.1,59.4](40/83例)であった。
副作用発現頻度は全投与例において90.7%(88/97例)であった。主な副作用は、悪心52.6%(51/97例)、下痢51.5%(50/97例)、嘔吐37.1%(36/97例)、貧血29.9%(29/97例)、血小板減少症20.6%(20/97例)等であった。[5.1、5.3参照]
注1)試験対象患者
・原発性骨髄線維症、及び真性多血症又は本態性血小板血症から移行した骨髄線維症患者(WHO分類及びIWG-MRT基準に基づき診断)
・DIPSSリスク分類22)の中間-1リスクで症状を伴う、中間-2リスク又は高リスクの患者(FEDR-MF-003試験及びJAKARTA2試験)
・IPSSリスク分類23)の中間-2リスク又は高リスクの患者(JAKARTA試験)
・ルキソリチニブに対して抵抗性又は不耐容と医師が判断した患者(JAKARTA2試験)
・造血幹細胞移植歴がない又は予定されていない患者(FEDR-MF-003試験)
・肋骨縁下に5cm以上の触知可能な脾腫(試験共通)又はMRI若しくはCTで脾臓容積が450cm3以上の脾腫(FEDR-MF-003試験のみ)を有する患者
・ベースラインの血小板数が5万/mm3以上かつ好中球数1000/mm3以上の患者
注2)1サイクルは28日間とした。
注3)本剤の承認された用法及び用量は、「通常、成人にはフェドラチニブとして1回400mgを1日1回経口投与する。なお、患者の状態により適宜減量する。」である。
注4)Last observation carried forward
18. 薬効薬理
18.1 作用機序
フェドラチニブは、ヤヌスキナーゼ(JAK)2に対する阻害作用を有する低分子化合物である。フェドラチニブは、JAK2の下流のシグナル伝達分子(STAT等)のリン酸化を阻害すること等により、腫瘍増殖抑制作用を示すと考えられている24)。
18.2 抗腫瘍作用
JAK2 V617F変異を導入した骨髄細胞を同所移植した骨髄増殖性腫瘍モデルマウスにフェドラチニブを反復投与したとき、白血球数、ヘマトクリット値及び脾臓重量の減少等が認められた(in vivo)25)。
19. 有効成分に関する理化学的知見
19.1. フェドラチニブ塩酸塩水和物
| 一般的名称 | フェドラチニブ塩酸塩水和物 |
|---|---|
| 一般的名称(欧名) | Fedratinib Hydrochloride Hydrate |
| 化学名 | N-tert-Butyl-3-[(5-methyl-2-{4-[2-(pyrrolidin-1-yl)ethoxy]anilino}pyrimidin-4-yl)amino]benzenesulfonamide dihydrochloride monohydrate |
| 分子式 | C27H36N6O3S・2HCl・H2O |
| 分子量 | 615.62 |
| 物理化学的性状 | 白色〜類白色の固体である。 |
| KEGG DRUG |
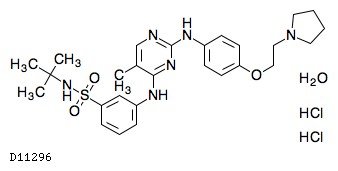
|
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
22. 包装
28カプセル[7カプセル(PTP)×4]
23. 主要文献
- 社内資料:ラット胚・胎児発生に関する用量設定試験(2025年6月24日承認、CTD 2.6.6.6.2.1)
- 社内資料:ラット胚・胎児発生に関する試験(2025年6月24日承認、CTD 2.6.6.6.2.2)
- 社内資料:国内第I/II相試験(FEDR-MF-003試験)(2025年6月24日承認、CTD 2.7.2.2.2.5.6)
- 社内資料:母集団薬物動態解析(2025年6月24日承認、CTD 2.7.2.2.2.5.7.2)
- 社内資料:食事の影響評価試験(2025年6月24日承認、CTD 2.7.1.2.1)
- 社内資料:母集団薬物動態解析(2025年6月24日承認、CTD 2.7.2.2.2.5.7.1)
- 社内資料:血漿タンパク結合試験(2025年6月24日承認、CTD 2.7.2.2.1.2)
- 社内資料:代謝酵素の同定(2025年6月24日承認、CTD 2.7.2.2.1.4)
- 社内資料:放射性標識体投与時の薬物動態及び代謝試験(2025年6月24日承認、CTD 2.7.2.2.2.1.2)
- 社内資料:腎機能障害の影響評価試験(2025年6月24日承認、CTD 2.7.2.2.2.3.1.1)
- 社内資料:肝機能障害の影響評価試験(2025年6月24日承認、CTD 2.7.2.2.2.3.2)
- 社内資料:ケトコナゾールとの薬物相互作用試験(2025年6月24日承認、CTD 2.7.2.2.2.4.1.1)
- 社内資料:生理学的薬物動態モデルによるシミュレーション(2025年6月24日承認、CTD 2.7.2.2.2.4.1.5)
- 社内資料:フルコナゾールとの薬物相互作用試験(2025年6月24日承認、CTD 2.7.2.2.2.4.1.4)
- 社内資料:リファンピシン及びエファビレンツとの薬物相互作用試験(2025年6月24日承認、CTD 2.7.2.2.2.4.1.3)
- 社内資料:ミダゾラム、オメプラゾール及びメトプロロールとの薬物相互作用試験(2025年6月24日承認、CTD 2.7.2.2.2.4.2.1)
- 社内資料:ジゴキシン、ロスバスタチン及びメトホルミンとの薬物相互作用試験(2025年6月24日承認、CTD 2.7.2.2.2.4.2.2)
- 社内資料:トランスポーター基質としてのフェドラチニブ(2025年6月24日承認、CTD 2.7.2.2.1.7)
- 社内資料:国内第I/II相試験(FEDR-MF-003試験)(2025年6月24日承認、CTD 2.7.6.5.3)
- 社内資料:海外第III相試験(EFC12153試験(JAKARTA試験))(2025年6月24日承認、CTD 2.7.6.5.1)
- 社内資料:海外第II相試験(ARD12181試験(JAKARTA2試験))(2025年6月24日承認、CTD 2.7.6.5.4)
- Passamonti F,et al., Blood., 115 (9), 1703-8, (2010) »PubMed
- Cervantes F,et al., Blood., 113 (13), 2895-901, (2009) »PubMed
- 社内資料:In vitro薬効薬理試験(2025年6月24日承認、CTD 2.6.2.2.1)
- 社内資料:In vivo薬効薬理試験(2025年6月24日承認、CTD 2.6.2.2.2)
24. 文献請求先及び問い合わせ先
文献請求先
レコルダティ・レア・ディジーズ・ジャパン株式会社
コンタクトセンター
〒107-0052
東京都港区赤坂4-8-18
電話:0120-108-100
製品情報問い合わせ先
レコルダティ・レア・ディジーズ・ジャパン株式会社
コンタクトセンター
〒107-0052
東京都港区赤坂4-8-18
電話:0120-108-100
25. 保険給付上の注意
本剤は新医薬品であるため、厚生労働省告示 第107号(平成18年3月6日付)に基づき、2027年5月末までは、投薬は1回14日間分を限度とされています。
26. 製造販売業者等
26.1 製造販売元
レコルダティ・レア・ディジーズ・ジャパン株式会社
東京都港区赤坂4-8-18